


“铜娃娃” | 了解肝豆状核变性的诊断和治疗
中国罕见病联盟
引言
肝豆状核变性(hepatolenticular degeneration, HLD)亦称Wilson病,是一种由于ATP7B基因缺陷导致编码的铜转运P型ATP酶功能缺陷或丧失,造成血清铜蓝蛋白合成减少以及胆道排铜障碍的常染色体隐性遗传代谢病,病理特征为铜离子在肝、脑、肾、骨关节、角膜等组织和脏器中过量沉积从而引起组织损伤,临床可见肝脏损害、神经精神表现、肾脏损害、骨关节病及角膜色素环(Kayser-Fleischer ring, K-F环)等。
肝豆状核变性患者有哪些临床表现?
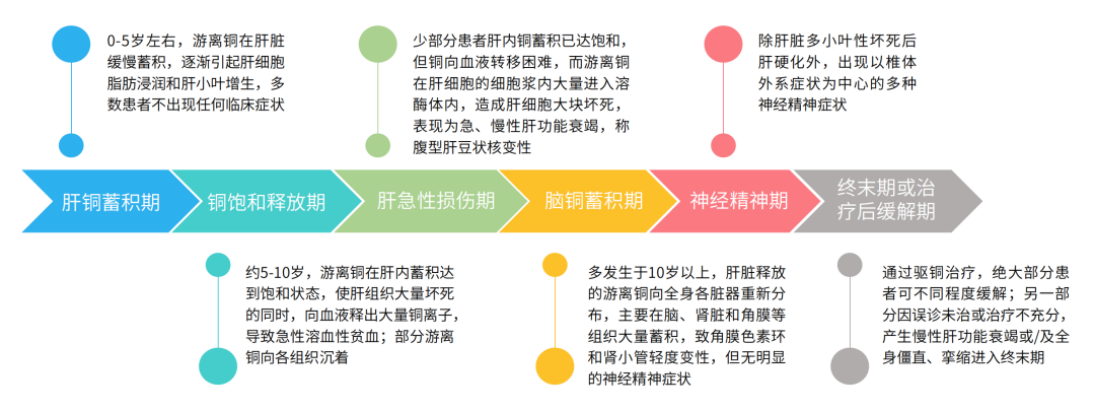
肝豆状核变性由于累及多系统、多器官,其临床表现差异较大,主要以肝脏病史或/及肝脏病征(79%)、锥体外系病征(97.6%)和角膜色素环(93%)为特征,按患者年龄组的病理改变可分为:I期(肝铜蓄积期)、II期a(铜饱和释放期)、II期b(肝急性损伤期)、III期(脑铜蓄积期)、IV期(神经精神期)、V期(终末期或治疗后缓解期)。
肝豆状核变性的检查与确诊
常规的检查包括:
(1)眼科查K-F环:角膜色素环(Kayser-Fleischer ring,K-F环)为角膜边缘的黄绿色或黄灰色色素环,一般在手电筒侧光照射下肉眼可见(7岁以下患者一般无法检出角膜K-F环)。
(2)血清铜蓝蛋白:铜蓝蛋白正常为200~500 mg/L,患者一般小于200 mg/L。铜蓝蛋白<80 mg/L 是诊断Wilson 病的强烈证据,若铜蓝蛋白<120 mg/L应引起高度重视,进行ATP7B基因检测明确诊断。
(3)24小时尿铜:收集24小时尿液可以了解尿中铜的含量。若成人≥100ug,儿童≥40ug,则需高度怀疑此病。
(4)肝脾检查:通常可见血清转氨酶、胆红素升高或白蛋白降低、肝脂质沉积、不规则结节、后期为肝硬化改变。
(5)其他检查:颅脑MRI检查(出现神经系统症状患者)、基因检测等。
遗传机制
肝豆状核变性是一种常染色体隐性遗传病,其致病基因ATP7B位于13q14.3区,编码一种P型铜转运ATP酶,该酶功能异常可导致人体铜代谢障碍,表现为铜经胆汁排泄障碍以及铜与铜蓝蛋白的结合率下降,过量的铜蓄积在肝、脑、肾、角膜等器官,出现相应的脏器功能损害。
目前有报道ATP7B基因致病变异多达900余种,不同人群中ATP7B基因变异的流行病学特征和常见变异位点均不同。欧洲WD患者人群中最常见的变异是p.H1069G,基因频率为13%~61%,亚洲人群的常见变异为p.R778L,基因频率为34%~38%,我国WD患者主要有3个高频致病变异,即p.R778L、p.P992L和p.T935M,占所有致病变异的50%~60%。
肝豆状核变性的诊断
WD可在任何年龄起病,早期诊断可改善WD患儿的预后,延误诊断是WD致死致残的主要原因,因此通过目前临床及基因手段,使WD在儿童期甚至婴幼儿期得到明确诊断可降低致残致死率。
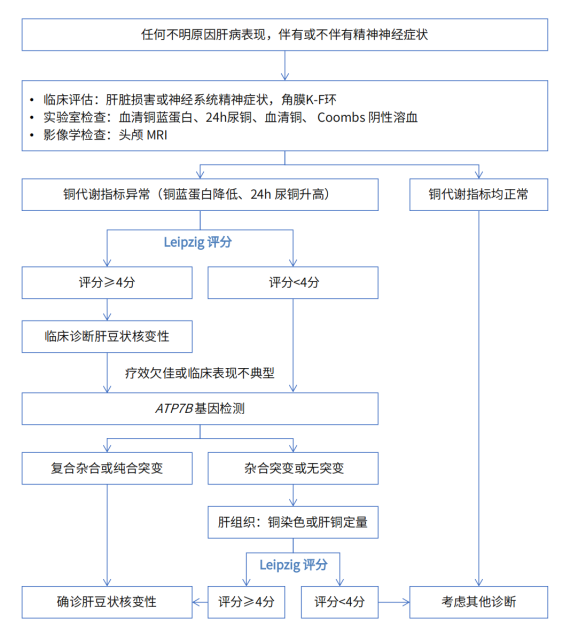
肝豆状核变性的诊断流程——《肝豆状核变性诊疗指南(2022年版)》
ATP7B基因变异以错义变异为主,主要为纯合变异以及复合杂合变异,少部分患者只找到单一杂合变异。对于临床表现不典型而又高度疑诊患者,可行ATP7B基因检测辅助诊断。
肝豆状核变性患者的治疗
肝豆状核变性主要以低铜饮食和药物治疗为主。患者应避免进食含铜量高的食物:如动物内脏、贝壳类、虾蟹类、坚果类、菌菇类、巧克力等。
目前国内常见的药物主要有两大类:
(1)青霉胺:青霉胺是一种螯合剂,可与体内沉积的铜结合,形成复合物,促进铜从尿中排泄。
(2)锌剂:常用于神经型患者或者无症状患者的一线治疗以及普通患者的维持治疗。锌剂诱导肠粘膜合成金属硫蛋白,阻止铜的吸收;诱导肝细胞合成金属硫蛋白,减轻肝脏铜的蓄积。对于Wilson病导致的急性肝衰竭或失代偿期肝硬化患者可能需要进行肝脏移植。Wilson病患者通常需要终身治疗,终身监测。
参考资料及文献
l 中华医学会神经病学分会神经遗传学组. 中国肝豆状核变性诊治指南2021[J]. 中华精神科杂志, 2021,054(004):310-319.
l European Association for Study of Liver (2012) J Hepatol 56,671
l 蓝秀健,李莹莹,等. 肝豆状核变性中的神经系统变性与铜毒性的综述. 逻辑学研究, (2007), 27(3),182-187.
l 胡智明,赵忠扩, 等. 肝豆状核变性治疗进展[J]. 肝胆胰外科杂志,2007, 19(3):3.
l Bennett J,Hahn SH.Clinical molecular diagnosis of Wilson'sdisease[J].Semin Liver Dis,2011,31(3):233-238.
l Cheng N,Wang H,Wu W,et al.Spectrum of ATP7B mutationsand genotypephenotype correlation in large - scale Chinesepatients with Wilson's Disease[J].Clin Genet,2017,92(1):69-79.
l Gomes A,Dedoussis GV.Geographic distribution of ATP7B mu-tations in Wilson's disease[J].Ann Hum Biol,2016,43(1):1-8.
l 中华医学会肝病学分会遗传代谢性肝病协作组. 肝豆状核变性诊疗指南(2022年版)[J]. 中华肝脏病杂志,2022,30(1):12.