


教学与科研 | 研究者发起的儿童罕见病临床研究合规风险管理——医疗机构视角
中国罕见病联盟
儿童罕见病领域存在巨大的未被满足的治疗需求,研究者发起的临床研究(IIT)为新药或新治疗策略的检验和发展提供了重要途径。但由于其受试者群体的特殊性、创新技术的应用以及组织形式的复杂性,这类研究在实施过程中面临立项审批、合同管理、数据保护、利益冲突管理等方面的诸多挑战和合规风险。本研究旨在分析儿童罕见病IIT的特殊性和挑战,梳理相关的法规和监管要求,为医疗机构提供合规风险管理的参考框架,以最大化IIT的益处。通过文献查阅和法律法规分析,结合实际工作经验及其他机构的实践框架,总结了儿童罕见病IIT在受试者特点、创新技术和组织形式方面的特殊性。在此基础上,提出了针对性的合规管理建议,包括建立风险评级和全周期风险监察机制、符合儿童受试者特点的知情同意和伦理审查机制、稳健的合同管理机制、全流程数据安全管理机制以及多学科团队和多渠道补偿机制。研究结论强调,医疗卫生机构、资助方及其他合作主体应结合IIT的特点,实施合规性管理,以保障研究的安全性和有效性,推动儿童罕见病治疗领域的创新与发展。
罕见病日益受到国家和社会各方的关注,WHO将罕见病定义为患病人数占总人口的0.65‰~1‰的疾病,但目前尚没有一个全球普遍接受的罕见病定义[1-2]。罕见病往往造成运动、神经、免疫等多系统渐进性损害,最终导致不可逆的后果。有研究显示,罕见病从发病到确诊平均需5年时间[3],且仅有1%的罕见病具有有效的治疗手段,因此,准确早期诊断和有效治疗是罕见病目前面临的重大挑战也是未满足的临床需求[4-5]。
研究者发起的临床研究(investigator initiated trial,IIT)指“医疗卫生机构开展的,以人类个体或群体(包括医疗健康信息)为研究对象,不以药品医疗器械(含体外诊断试剂)等产品注册为目的,研究疾病的诊断、治疗、康复、预后、病因、预防及健康维护等的活动”[6]。IIT通常致力于探讨具有创新潜力的医学概念,研究药物在新的患者群体或新的适应证中的疗效和安全性,尽管制药企业发起的临床试验仍是新药注册评审的主要依据,但高质量的IIT在一定情况下,可作为药品注册或增加适应证、临床应用指导原则与诊疗指南修订的重要参考[7],因此IIT逐渐成为儿童罕见病领域制药行业与独立研究者(包括个人、学术机构和协作团体)之间合作的基石。
由于中国儿童罕见疾病IIT起步较晚,仍存在相应法规指引不够明晰、可参考的先例缺乏、涉及儿童和罕见病患者这一独特身心和社会经济状况的受试群体情况,并且罕见病更多采用基因治疗、细胞治疗等新的手段,医疗卫生机构作为IIT研究实施的责任主体,面临复杂的伦理审查和合规管理挑战[8]。本文通过文献综述,在CNKI、万方、PubMed数据库,以“罕见病”“研究者发起临床研究”“风险管理”“合规”等为关键词检索文献,纳入实证研究与系统综述,分析儿童罕见病IIT的特点和挑战,梳理中国对于儿童罕见病IIT相关的法规和监管要求,基于实际工作经验,从医疗机构的角度,探讨如何在最大化IIT益处的同时进行有效的合规风险管理和控制。
1儿童罕见病治疗领域的研究者发起的临床研究的特殊性
1.1儿童罕见病受试者群体具有特殊的身心特点及社会经济状况
首先,“儿童不是缩小版的成人”。研究者不能简单地降低或缩减成人的治疗方案以适合儿童,因为未成年人的身体成分和器官成熟度在数量和质量上与成人不同[9]。儿童正处于生长发育期的动态变化的过程中,对药物、医疗新技术等干预措施更加敏感,常常表现出耐受性差及不良反应率偏高,不同年龄的儿童体内药物代谢动力学和药效动力学也存在差异。其次,儿童容易出现恐惧、疼痛、与父母分离和焦虑等心理问题,而青春期受试者容易出现各种心理冲突和矛盾,呈现出敏感、脆弱的心理状态。再次,罕见病儿童的监护人需要照顾患儿而放弃工作,治疗费用较高,且往往需要跨省市就医[10],因此家庭收入水平较低且诊疗经济负担较重[11],被认为是“脆弱人群中的弱势群体”。最后,儿童监护人对儿童参与临床研究理解不足或者对新的治疗手段的风险的担忧,有可能造成监护人阻碍儿童参与临床研究。
1.2儿童罕见病IIT研究规模小,单臂研究多
由于罕见病发病率低、患者少、临床异质性高,难以开展随机对照研究,往往是小规模的单臂研究。近10年中国开展的罕见病临床试验中,48.3%为单臂设计,有些试验受试者不足10例[10]。
1.3儿童罕见病IIT前期的数据结果可能不够充分
尽管临床研究有“先成人后儿童”的原则,但某些罕见病中儿童治疗需求强烈,有些IIT可能首先就在儿童或婴儿中开展[12]。
1.4儿童罕见病的IIT常涉及创新药/疗法的开发
小分子药物治疗、抗体治疗、蛋白质替代治疗、基因治疗、造血干细胞治疗和药物再利用等是目前罕见疾病的主要治疗策略[13]。这类探索、创新治疗可预测性不强、临床风险较高,对儿童受试者带来的风险和损害的可能性更较大。
1.5儿童罕见病IIT涉及人类遗传资源管理
人类遗传资源材料和人类遗传资源信息,即含有人体基因组、基因等遗传物质的器官、组织、细胞等遗传材料,以及利用人类遗传资源材料产生的数据等信息资料。由于遗传缺陷是导致罕见病的重要因素,大约80%的罕见病由单基因缺陷导致[4],罕见病的IIT常常涉及人类遗传资源材料和信息的收集、储存和管理。
1.6儿童罕见病IIT涉及更为复杂的组织合作关系
尽管IIT的责任主体并非制药企业,但是儿童罕见病IIT通常涉及创新的生物制剂,这些创新生物治疗的研究物资往往由医药生物企业资助,涉及资助方与研究者之间的利益冲突,也考验着研究者研究方案的设计水平;此外,罕见病患者数量稀少,常常需要开展多中心临床研究,涉及牵头单位与分中心的权利义务分配问题。
2开展儿童罕见病研究者发起的临床研究合规风险点
2.1立项管理合规风险
根据《医疗卫生机构开展研究者发起的临床研究管理办法》[6],医疗卫生机构及研究者作为发起人,应当遵守有关法律法规、部门规章及有关规范性文件和技术准则、伦理规范的要求,按规定完成立项审查、科学性审查、伦理审查等程序,并且有关信息应当在国家医学研究登记备案信息系统按要求完成上传。如未能完成必要立项程序,而擅自启动临床研究并招募患者,将可能受到行政处罚并引发纠纷。立项管理的另一个合规风险在于,在涉及超范围的IIT中,《医疗卫生机构开展研究者发起的临床研究管理办法》中未对超范围研究的性质判定技术标准与流程、立项审查做明确规定。由于不同医疗机构的临床研究审批管理标准不同,在无统一标准的情况下,难免遇到概念模糊、判断不清的情形,有些机构可能对儿童罕见病IIT的审查过于保守,有些则可能则过于冒进。
2.2伦理审查合规风险
《涉及人的生命科学和医学研究伦理审查办法》[14]中,对涉及弱势群体的临床研究强调了“应予以特别关注”。涉及儿童的医学研究须遵循风险最小化原则,即研究风险不得超过其日常医疗风险,除非潜在收益显著且无更低风险替代方案[15]。罕见病家庭经济负担重,如未能进行科学且充分的受试者风险-受益评估,高额补偿(如覆盖医疗费用)可能被视为变相诱导。儿童罕见病IIT的知情同意也因其研究主体(儿童)和研究对象(罕见病)的双重特殊性而面临多重复杂性,一方面由于罕见病知识高度专业化导致的沟通挑战和治疗误解,另一方面罕见病儿童理解能力的差异较大,是否需要执行儿童版知情同意难以明确界定。
2.3个人隐私保护、数据安全及人类遗传资源管理合规风险
根据《中华人民共和国数据安全法》《中华人民共和国个人信息保护法》《个人信息跨境处理活动安全认证规范》《人口健康信息管理办法(试行)》等相关数据保护规范,IIT涉及包含受试者个人身份信息、统计信息、通信信息等的个人属性数据,以及包含受试者主诉、现病史、检验检查数据等的健康状况数据、包含病历资料、检查报告、病程记录等的医疗应用数据、包含受试者医疗交易信息、保险信息在内的医疗支付数据、医疗机构运营数据等的卫生资源数据。在儿童罕见病IIT中,容易忽视资助方、医疗机构和第三方机构各自在个体信息处理中的权利和义务,缺乏数据安全计划及措施,未能注意受试儿童及其监护人个体信息的去标识化,有些项目可能存在过度收集的问题,以及缺少个人信息返还和删除的有关约定和告知。
对于基因和细胞治疗等创新治疗IIT项目,经常涉及人类遗传资源材料和信息,根据《中华人民共和国生物安全法》《中华人民共和国人类遗传资源管理条例》,人类遗传资源的获取、利用需要国家和个人的双重同意。儿童人类遗传资源较成人更为稀缺,其研发投入更大、周期更长,导致了其获取与使用的难度加大、风险增高,更加不利于权益保护,也更易造成人类遗传资源的流失。
2.4合同管理风险
儿童罕见病IIT研究可能涉及多个主体,包括研究者、医疗机构、药企、第三方基金会或学会等,不同主体的资质和能力参差不齐,增加了合同管理的复杂性和风险。IIT研究中较常出现4类研究协议,具体如下:①委托/资助方和医疗机构/研究者签署两方合同;②资助方、合同研究组织(contract research organization,CRO)与医疗机构签署三方合同;③CRO与医疗机构签署两方合同;④牵头单位与分中心单位形成的合作合同关系。医疗机构最为利益相关的是第一类合同关系,其主要争议点在于受试者不良反应处理流程和赔偿责任承担、知识产权条款;而较为容易被临床研究者忽略的是数据安全与个人信息保护条款、商品/器械供应和配送细节条款。对于受试者不良反应处理流程、保险和赔偿责任承担,《医疗卫生机构开展研究者发起的临床研究管理办法》[6]要求“医疗卫生机构应当建立受试者争议和投诉的处理机制,科学判定是否有损害及其产生的原因,合理划分责任,按照约定或有关管理规定,对受到损害的受试者进行合理的补偿或赔偿”,但并未对资助企业和医疗机构的补偿或赔偿责任做一个明确的建议。在具体的儿童罕见病IIT项目中,不同的资助企业对不良事件的赔偿原则不同。由于儿童罕见病研究的高风险性,各方可能在风险分担上存在分歧,有些企业明确只提供药品而不承担任何赔偿责任,或者仅由企业资助购买临床研究保险,但是对于保险未能覆盖的损害赔偿(例如起付线以下、超过保额范围、超过保险覆盖期限)如何划分责任,各方在合同中未能明确,导致合同签订困难或成为儿童受试者和家属、医疗机构和资助企业日后争议的焦点。
2.5药企资助的合规性风险
对于涉及企业资助的儿童罕见病IIT的另一个风险点在于企业和医疗机构或者研究人员之间的利益冲突问题,国家市场监督管理总局发布的《医药企业防范商业贿赂风险合规指引(征求意见稿)》[16]指出 “禁止医药企业通过设立虚假项目或者假借临床研究名义,向临床试验机构及研究者输送不当利益,以换取在医药产品入院、推广或者销售等方面的竞争优势或者交易机会。禁止医药企业直接或者通过第三方服务机构向临床试验机构或者研究者个人输送不当利益,以换取加快、推进临床研究或者操纵临床研究数据,使临床研究结果有利于本医药企业产品”。为了避免商业贿赂风险,一些医疗机构不接受药企直接资助,而是通过第三方基金或者学会获得资助。而这些通过第三方居间的合规成本,既无法做到审计层面的无痕,又增加了沟通和费用的双重成本。
3研究者发起的儿童罕见病临床研究的合规风险管理建议
3.1将儿童罕见病IIT纳入中高风险IIT并建立全周期风险监察机制
IIT项目风险评级是指根据项目的各因素进行风险赋值、统计、建模和评级。风险评级因素通常包括受试人群风险(纳入病情较重的研究者风险大于入组病情较轻的患者)、研究设计(如干预性研究风险大于观察性研究)、干预措施(药物、药械、手术方式、心理)、作用机制(替代治疗或非替代治疗)、作用靶点(全新靶点或已研究的靶点)、研究者经验、研究中心数、样本量、协调员配比、资助及合作方式等。根据得分情况,可将项目划分为不同的风险等级(高风险、中风险、低风险等)。儿童罕见病IIT由于疾病情况较重、治疗风险较高,且单臂研究类型较多,往往需要开展多中心研究、创新治疗涉及药企资助合作,应纳入中高风险的IIT进行管理、并建立全周期的风险监察机制。
借鉴美国食品药品监督管理局风险监察机制,基于项目的风险级别,设定需要进行数据识别的关键流程节点、设定监测频率、建立风险管理计划、风险管理报告,以及设定触发现场检查的指标评分和阈值等[17-18]。针对儿童罕见病IIT应制订详细的风险管理计划、建立项目风险管理报告、梳理实施过程的关键流程节点,设立独立数据监察委员会定期审查数据安全性,重要指标的构成比、离群值等,对风险进行追踪评估,调整监察计划的时间、评估和内容等。
3.2建立符合儿童罕见病受试者特点的知情同意和伦理审查规程
儿童罕见病IIT的知情同意过程,应遵循《涉及人的生命科学和医学研究伦理审查办法》[14]第三十四条的规定,充分体现对儿童的保护,可以根据年龄和成熟度分阶梯的知情同意/同意过程[19],比如年幼的儿童,可以设计互动式同意材料(如增强现实虚拟实验室参观、选择题形式的风险认知测试)。合法监护人的受教育程度也存在差异,研究者应耐心沟通,尽可能少地使用专业术语来解释研究信息,使用的话语尽量做到简单易懂,灵活地采用适当的解释方法。充分履行告知义务,最后达到使儿童及其合法监护人完全理解自主作出决定。
在儿童罕见病的IIT伦理审查方面,要重点关注以下几个方面:①风险最小化。研究风险不得超过儿童日常医疗或疾病自然进展的风险,除非研究具有直接治疗潜力且无更低风险替代方案。尽量明确量化或定性描述其潜在收益(如延缓病程、改善生活质量),确保其高于风险。②如果研究设计中涉及小样本、单臂试验等方法。伦理审查应关注是否有充分的评估和证据支持开展小样本研究、是否符合《罕见疾病药物临床研发技术指导原则》[20]中对开展单臂试验的基本条件。③隐私保护。特别是在涉及遗传信息的研究中,防止可能的歧视或社会偏见。④保障措施。确保试验过程中有完善的监护机制,及时处理不良事件,保障儿童罕见病受试者的安全和权益,给予儿童罕见病患者以比成年受试者更长期的后续关怀。
3.3建立稳健严谨的儿童罕见病IIT合同管理机制
参考健康研究促进发展委员会(Council in Health Research for Development,COHRED)[21]关于研究合作合约谈判指南,医疗机构应建立IIT的合同模板或标准检查清单,这些模板应包括允许“合理灵活性”的条款,并作为与外部资助机构谈判的起点。对于与其他研究合作方(如保险政策、数据和材料传输协议等)的谈判,同样可以采用模板和指导作为基础。关键组成部分如表1所示。通过制定全面的研究协议,所有相关方可以在研究开展过程中保持透明性和合作性,从而确保项目的成功并减少潜在的争议和风险。
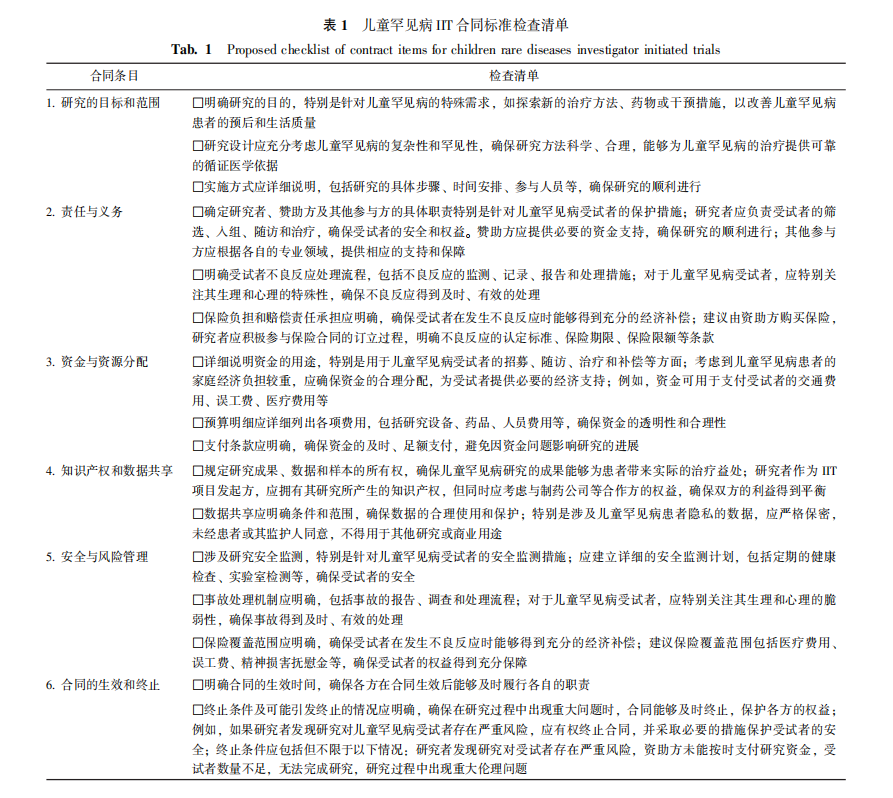
知识产权管理方面。总体而言,研究者作为儿童罕见病IIT项目发起方,当然拥有其研究所产生的知识产权。IIT中,绝大多数制药公司出于未来商业利益的考虑,会重点关注IIT试验研究中所产生研究数据的所有权,以及协议履行过程中所产生的技术成果及所有知识产权成果的归属问题。具体实操中如何划分知识产权的归属,取决于双方的协商结果。在处理这些问题上,建议研究者及其所属的医疗机构仔细审查这些条款,必要时联合研究者、相关研究科室、法务人员进行合同谈判,以确保其权益得到充分保障。
对于受试者不良反应的处理流程、保险和赔偿责任承担,医疗卫生机构应当全面梳理该IIT可能发生的不良事件、对应的治疗和相应的补偿和赔偿方案,建立受试者和研究对象损害风险预防、控制及财务保障机制。如涉及购买保险,需明确保险的购买方是资助方还是发起方,即使由资助方购买保险,研究者也须积极参与资助方与保险公司的业务交涉及保险合同的订立过程,明确其不良反应的认定标准,查看相关免责条款,如保险期限、保险限额等。如经研究者和研究科室研判属于高风险的研究项目,应采取提议提高受试者保费、提高每次事故赔偿限额或延长承保期限等措施,以规避医疗机构在此情形下的赔偿风险。
3.4建立权责明晰的全流程数据管理机制
医疗机构应当根据《中华人民共和国个人信息保护法》《人口健康信息管理办法(试行)》等数据保护规范,建立IIT项目相关的数据保密和保护机制、处理程序及救济措施,在数据采集、传输和存储的过程中保证安全性、完整性、保密性和可追溯性等,做好去标识化管理,加强信息的“脱敏”。在临床研究协议中,明确各方在个体信息处理中的权利和义务、个人信息删除、返还约定、禁止转委托条款等。对于原始医疗记录的衍生文件及数据(包括但不限于病例报告表)应仅限于本研究使用,不得用于其他延展研究及扩展性研究。对于患者隐私信息,医疗机构仅在相关知情同意、授权和备案的范围内使用,合作方应承诺不会接触、处理以及获取受试者的个人信息。另外,建议医疗机构与其他合作方另行签署保密协议,保密期限应当覆盖到临床试验研究结束后的5年以上,并约定违约赔偿及守约方救济等问题。
对于罕见病IIT尤其需要注意的是,中国已有的活动规律的罕见病病友组织130家[22],IIT常常通过罕见疾病患者组织、患者倡导者、患者小组或患者线上社区寻找患者。在考虑与某病种患者组织合作或进行访谈时,应注意上述数据合规要求。
此外,IIT研究必须梳理项目过程中是否包含人类遗传资源,如全血、血清、血浆、组织切片等包括含有人体基因组、基因等遗传物质的器官、组织、细胞等遗传材料,并且需要严格排查各合作方,包括分中心、资助方、第三方检测机构或资助方聘请的第三方检测实验室是否存在外资背景。明确所涉人类遗传资源的使用环节是否涉及审批或备案流程、是否涉及国际合作。如涉及人类遗传资源材料出境,或者为取得相关药品和医疗器械在我国上市许可的临床试验涉及的探索性研究部分,则应当申请人类遗传资源国际科学研究合作行政许可。这也意味着,IIT研究项目中的任何一方均不得超出备案范围采集、收集或使用研究所涉及的人类遗传资源材料。
3.5建设儿童罕见病IIT多学科团队及多渠道补偿机制
儿童罕见病等高风险IIT应有多学科团队的支持保障。除了临床研究设计方案中所要求的专业知识和经验,成员中还应包括具备研究监察、临床协调、合规、数据管理、质量控制的专职人员。对于儿童罕见病IIT,也可以建立特殊的多渠道风险补偿和赔偿机制,资金的来源可以是社会、政府、企业等多渠道,为受试者提供额外的安全网,同时,这也需要政府、医疗机构、研究者和社会各界共同努力,制定合理的管理规则和运作机制,确保经费的透明度和公平性,在保护受试者权益和推动医学进步之间找到平衡。这不仅有助于提高临床研究的质量,也有助于增强公众对医疗机构临床研究的信任和参与度。
综上所述,儿童罕见病领域存在巨大的未被满足的治疗需求,IIT有助于新药或新治疗策略的检验和发展。医疗卫生机构、资助方以及其他合作主体在开展临床研究过程中,应当不断完善各项合规管理工作,遵循相应的伦理原则,协调各方利益,建立适合儿童罕见病IIT特色的风险管理制度、方案和流程等,保障儿童罕见病IIT安全合规实施,促进创新转化,为中国儿童健康事业创新发展贡献力量。
作者贡献:张婧绮负责文章撰写和文献检索、筛选、分类整理,建立文献数据库,协调修改意见,整合多方反馈;左连东负责文章撰写,追踪领域内最新研究进展,补充关键制度、逻辑优化及学术规范性审核;高雪琪负责文章撰写,补充关键法律法规;斯文越负责追踪领域内最新研究进展;罗睿参与图表设计,补充材料整理;吴强负责全文语言润色;周文浩主导主题的确定、关键研究方向的分析与归纳。
利益冲突:所有作者均声明不存在利益冲突。
参考文献
[1]人民网. 《中国罕见病定义研究报告2021》发布[EB/OL]. (2021-09-13)[2024-11-30]. health.people.com.cn/n1/2021/0913/c14739-32225468.html.
[2]李定国,王琳,许小幸. 从临床流行病学角度思考中国罕见病定义修订[J].临床儿科杂志,2021,39(8):561-564.
[3]Dwyer AA, Uveges MK, Dockray S, et al. Exploring rare disease patient attitudes and beliefs regarding genetic testing: implications for person-centered care[J]. J Pers Med, 2022,12(3):477.
[4]左玮,张波,张抒扬.罕见病用药保障现状与挑战[J].罕见病研究,2024,3(2):155-163.
[5]World Health Organization. The selection and use of essential medicines: report of the WHO Expert Committee, 2023 (Including the 23th WHO Model List of Essential Medicines and the 9th WHO Model List of Essential Medicines for Children)[EB/OL]. (2023-04-24) [2024-05-21]. https://iris.who.int/bitstream/handle/10665/ 189763/9789241209946_eng.pdf?sequence=1.
[6]国家卫生健康委员会,国家中医药局,国家疾控局. 关于印发医疗卫生机构开展研究者发起的临床研究管理办法的通知(国卫科教发〔2024〕32号)[EB/OL]. (2024-09-18) [2024-10-20].http://www.nhc.gov.cn/qjjys/s7945/202409/bdb18f33eea8462b876c155d5ba529c4. shtml.
[7]李园园,胡岚岚,王砚,等. 研究者发起的超范围临床研究管理策略探析[J]. 中华医学科研管理杂志,2024,37(1):66-69.
[8]叶朝辉,严荧燕,朱卓儿,等. 基于药品管理探讨研究者发起的儿科药物临床研究实施风险及对策[J]. 中华医学科研管理杂志,2024,37(5):424-428.
[9]Cuzzolin L, Atzei A, Fanos V. Off-label and unlicensed prescribing for newborns and children in different settings: a review of the literature and a consideration about drug safety[J]. Expert Opin Drug Saf, 2006, 5(5):703-718.
[10]陈晨,韩晓红.中国罕见病药物临床试验10年现状分析:基于《第一批罕见病目录》[J].协和医学杂志,2022,13(6):1028-1035.
[11]白桦,张抒扬.罕见病临床研究中的伦理问题[J].临床药物治疗杂志,2019,17(11):14-17,23.
[12]中国临床试验注册中心. GC301腺相关病毒注射液基因治疗>1岁婴儿型庞贝病患者安全性、耐受性及初步疗效的临床探索试验[EB/OL]. (2023-05-06)[2024-09-30]. https://www.chictr.org.cn/showproj.html?proj=180547.
[13]Tambuyzer E, Vandendriessche B, Austin CP, et al. Therapies for rare diseases: therapeutic modalities, progress and challenges ahead[J]. Nat Rev Drug Discov, 2020, 19(2):93-111.
[14]国家卫生健康委员会,教育部,科技部,等. 关于印发涉及人的生命科学和医学研究伦理审查办法的通知(国卫科教发〔2023〕4号)[EB/OL]. (2023-02-18) [2024-12-01].https://www.gov.cn/zhengce/zhengceku/2023-02/28/content_5743658.htm.
[15]郭春彦,张怡,丁倩,等.涉及儿童的临床研究伦理审查指南(北京)[J].中国临床药理学杂志,2021,37(11):1476-1480.
[16]国家市场监督管理总局. 市场监管总局关于公开征求《医药企业防范商业贿赂风险合规指引(征求意见稿)》意见的公告[EB/OL]. (2024-10-11) [2024-11-01].https://www.samr.gov.cn/hd/zjdc/art/2024/art_8ff267f2f83e49af9754a937f841f37a.html.
[17]U.S. Department of Health and Human Services Food and Drug Administration. Oversight of clinical investigations-a risk-based approach to monitoring[EB/OL].(2013)[2022-11-15]. https://www.fda.gov/regulatory-information/ sea-rch-fda-guidance-documents/oversight-clinical-investigations-risk-based-approach-monitoring.
[18]袁宝石,王胤凯,倪如月,等. 基于风险监查策略在研究者发起的临床研究中思考与实践[J]. 中华医学科研管理杂志,2023,36(3):182-188.
[19]European Union. Ethical considerations for clinical trials on medicinal products conducted with the paediatric population [J]. Eur J Health Law, 2008,15(2):223-250.
[20]国家药品监督管理局药品审评中心. 国家药监局药审中心关于发布《罕见疾病药物临床研发技术指导原则》的通告(2021年第71号)[EB/OL]. (2022-01-06)[2024-12-14]. https://www.cde.org.cn/main/news/view InfoCommon/c4e1ef312a0a0c039a7a4ca55b91d4e8.
[21]Marais D. Where there is no lawyer: Guidance for fairer contract negotiation in collaborative research partnerships. Council in Health Research for Development (COHRED)[EB/OL] (2013)[2024-12-01]. http://cohred.org/wp-content/uploads/2012/04/ Fair-Research-Contracting-Gui-dance-Booklet-e-version.pdf.
[22]郝婵娟,倪鑫.儿童罕见病诊治现状及展望[J].罕见病研究,2022,1(3):229-232.