


综述 | 数智化技术在溶酶体贮积病辅助诊断中的应用现状
中国罕见病联盟
目的系统梳理数智化技术在溶酶体贮积病辅助诊断中的应用现状,探讨面临的机遇和挑战,以及未来发展趋势,以期为数智化驱动辅助诊断技术的发展提供参考和建议。方法通过检索PubMed、Web of Science、Embase、中国知网、万方数据知识服务平台、维普中文科技期刊数据库等中英文数据库,纳入关于数智化技术在溶酶体贮积病诊断中应用的研究,定性分析其方法和结果,并根据类别归纳总结数智化技术在溶酶体贮积病辅助诊断中的应用现状,以及存在的不足和挑战。结果数智化技术在溶酶体贮积病的早期筛查和诊断中具有巨大潜力,尤其是大数据存储及管理技术、大数据挖掘及分析技术、机器学习、自然语言处理和计算机视觉等数智化技术,可以识别潜在患者、发现新的生物标志物、识别并定量分析溶酶体贮积病症状、探索基因与溶酶体贮积病之间的关系等,提高诊断效率和准确性。结论数智化技术能够提高早期诊断的准确性,在溶酶体贮积病的诊断研究中具有广阔的应用前景。未来应在DI-HEALTH理论框架指导下,构建全方位、多层次、立体化的溶酶体贮积病辅助诊断体系和技术。
溶酶体贮积病(lysosomal storage disease,LSD)是一种罕见的遗传代谢性疾病,因基因突变导致细胞溶酶体内大分子不能或仅部分被代谢清除,致使细胞功能障碍,进而出现神经退行性改变、免疫功能下降、骨骼畸形、生长发育迟缓等症状,如不及时干预,疾病将快速进展,并威胁生命。常见的LSD包括黏多糖贮积症、黏脂贮积症、神经鞘脂贮积症(法布雷病、戈谢病、尼曼匹克病、Tay-Sachs病)等[1]。早期诊断并及时干预是延缓疾病进展、控制并发症发生风险、改善患者预后的关键措施[1]。然而,多种因素却制约着LSD的早期准确诊断。首先,LSD是一种多器官、多系统受累的疾病[2],临床表现的多样性和复杂性显著增加了诊断难度[3];其次,疾病随年龄进行性加重的特点使早期症状相对隐匿,易造成漏诊或误诊[4-5];最后,疾病的罕见性使临床医生缺乏对疾病的敏感度[6-7],极大地降低了早期诊断率。
“数智化”一词最早由北京大学“知本财团”课题组于2015年正式提出,为数字智能化与智能数字化的结合[8],可以用“数字化+智能化”来定义和解释[8-9]。数智化技术是数据技术和智能技术相结合的概念[10],因其能基于海量多模态医疗数据构建疾病预测模型,并对模型进行持续优化,近年来逐渐被用于疾病的辅助诊断。与传统辅助诊断方法相比,基于数智化技术的辅助诊断方法具有效率高、准确性高、成本低、非侵入性等特点。目前,该技术已广泛应用于常见病的诊疗工作,并取得积极的效果。例如,在影像诊断中应用计算机辅助的半自动化定量评估系统,显著提高了诊断结果的客观性与可重复性[11-12];利用深度学习卷积神经网络技术解读心电图,可快速识别一些人工难以发现的心电信号,提高心血管疾病的诊断准确性[13]。虽然数智化技术在LSD诊断中也有探索性实践,但距离大规模应用仍存在较大差距。同时,目前仍缺乏对数智化技术辅助LSD诊断的应用场景、应用案例和应用效果的系统化归纳总结。因此,本研究利用范围综述的方法,系统梳理数智化技术在LSD诊断中的应用现状、面临的机遇和挑战,以及未来发展趋势,以期为基于数智化技术的LSD辅助诊断的应用推广和持续优化提供参考与建议。
1资料与方法
1.1文献检索
检索PubMed、Web of Science、Medline、Embase、中国知网、万方数据知识服务平台、维普中文科技期刊数据库等中英文数据库,检索范围为标题、摘要、主题词、关键词等常用字段。检索词包括“溶酶体贮积病”(“Lysosomal Storage Diseases”等)、“数智化”("Artificial Intelligence""Decision Support Techniques""Medical Informatics Computing""Big Data""Data Mining""Information Storage and Retrieval""Machine Learning""Deep Learning""Natural Language Processing""Data Visualization""Digital Health""Digital Technology"等)、“诊断”(“diagnosis”)等。
1.2纳入排除标准
纳入标准:①研究主题为基于数智化技术的疾病辅助诊断;②研究疾病为溶酶体贮积病;③对诊断效果进行明确评价;④公开发表的研究。排除标准:①非中、英文文献;②重复发表的文献;③会议论文及摘要、述评、综述、研究方案等文献类型;④无法获取全文的文献。
1.3文献筛选与信息提取
由两名研究者分别开展文献筛选和信息提取工作。首先将文献导入文献管理软件Zotero进行去重;其次,阅读标题和摘要进行初筛;最后,阅读文献全文进行二次筛选,确定最终纳入的文献。对意见不一致的文献由第三位研究者独立评价后共同讨论决定是否纳入。从文献中提取的信息包括:作者、发表年份、地区、研究人群、数据来源、研究疾病、数智化技术类型、研究内容、应用效果。采用定性分析方法对提取的信息进行汇总分析。
去重后共获得相关文献233篇,经逐层筛选后,最终纳入24篇研究,文献筛选流程和结果见图1。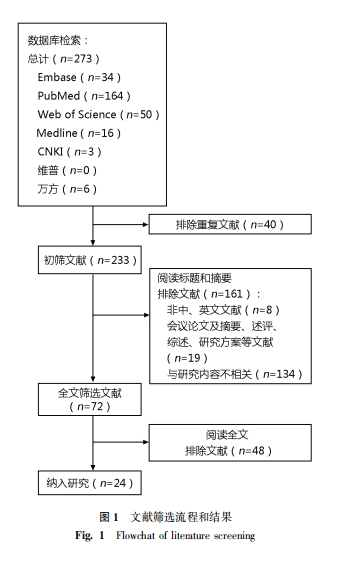
2结果
2.1纳入文献基本特征
表1为纳入文献的基本特征。文献发表年份为2010—2024年,来自全球6个国家。研究的病种包括黏多糖贮积症(n=7)、法布雷病(n=7)、戈谢病(n=4)、庞贝病(n=2)、黏脂贮积症(n=2)、α-甘露糖贮积症(n=1)、尼曼匹克病(n=1)、辛德勒病(n=1)。涉及的数智化技术包括大数据存储及管理技术(n=2)、大数据挖掘及分析技术(n=4)、机器学习(n=10)、自然语言处理(n=1)、计算机视觉(n=7)。

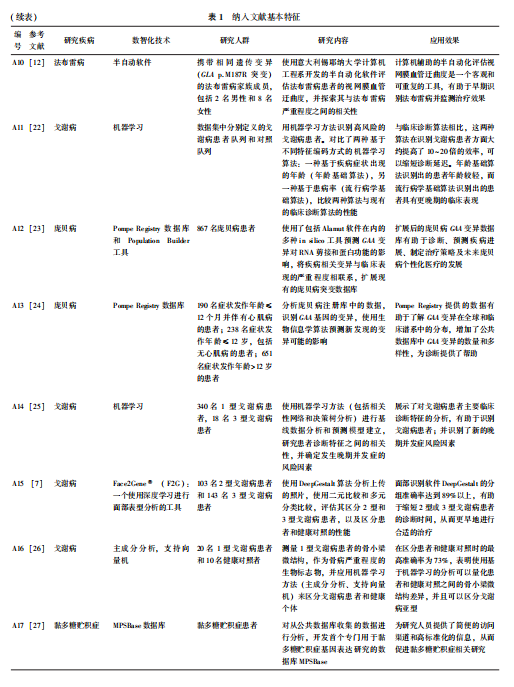
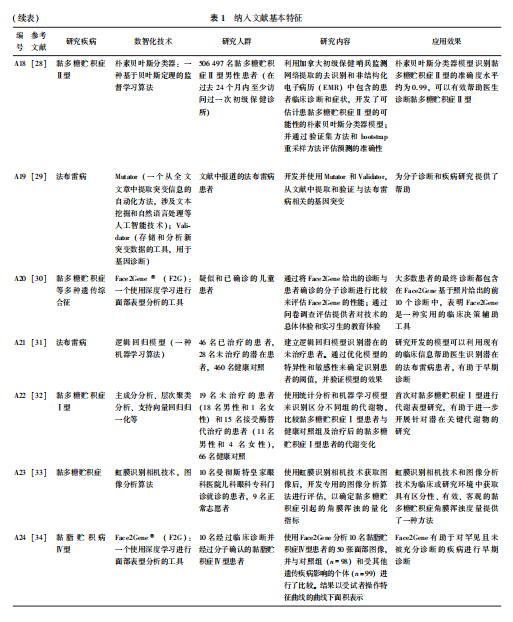
2.2数智化技术在溶酶体贮积病诊断中的应用及效果
对纳入文献进行归纳总结后,根据常见的数智化技术类别,可将涉及的数智化技术分为:大数据存储及管理技术(如构建患者信息数据库、突变基因信息数据库等)、大数据挖掘及分析技术(如数据挖掘技术等基于数据库的算法)、机器学习、自然语言处理和计算机视觉(如利用计算机进行表型和特征的定性和定量分析等)。
2.2.1大数据存储及管理技术
大数据存储与管理是指对结构化、半结构化或非结构化的多模态数据进行解析、融合与存储,并实现融合数据的快速查询与分析[35],进而支持数据驱动的辅助诊断决策。
Riise Stensland等[17]构建了来自全球41个国家和地区的α-甘露糖苷症患者数据库,包含140种MAN2B1基因突变位点和每名患者的临床表型、实验室检查等数据,实现了α-甘露糖苷症基因型和表型的精准匹配。
Soares等[27]开发了首个用于分析黏多糖贮积症基因型的数据库MPSbase,整合了超过50 000个用于描述基因功能的富集术语和92 000个基因信息,帮助研究者利用高通量测序数据查询不同亚型黏多糖贮积症患者的差异化基因表达,为遗传诊断提供支持。
2.2.2大数据挖掘及分析技术
利用大数据挖掘和分析技术可以帮助研究者在海量医学健康数据中提取、加工、分析与疾病诊断相关的内容,并助力疾病机制的探索和验证。
学者Nio等[23]和Reuser等[24]使用大数据挖掘技术分别对全球最大的庞贝病数据库进行深度分析,新发现多个可致病的酸性α-葡萄糖苷酶基因突变位点,并预测了新发变异位点对疾病发生的影响。研究结果不仅扩展了Pompe Registry数据库,更为庞贝病的诊断提供了新证据。学者Moynihan等[20]利用大数据挖掘和分析技术对128万人的电子健康档案进行重新分析,在原有4例法布雷病患者的基础上,新发现了2例确诊患者,证实了大数据驱动的疾病诊断的可行性。
传统的基因测序数据分析与解读方法非常耗时且成本较高,学者Ghasemi等[21]利用大数据算法构建了一种新型测序数据分析方法——NetEM。使用该方法分析LSD患者基因测序数据、识别致病基因的效果显著优于当前使用的3种方法(Cardigan、DIAMOnD和GeneWanderer)。
2.2.3机器学习
因对不同数据分布特征的包容性和持续学习与优化的特性,机器学习算法被广泛用于临床数据深度挖掘和疾病诊断预测。4项研究[15,18,26,32]利用不同的机器学习方法对数据进行深度分析,发现了具有较高诊断意义的生物标志物。Moreno-Barea等[18]利用数据增强技术提高C型尼曼匹克病尿液样本数据集的预测能力,并识别新的生物标志物,基于该标志物的疾病预测灵敏度提高了20%~50%。Tebani等[15]运用无监督机器学习方法——主成分分析和层次聚类分析发现了法布雷病的4种具有鉴别诊断意义的蛋白质,包括成纤维细胞生长因子2、血管内皮生长因子A、血管内皮生长因子C和白细胞介素-7;此外,还使用类似的机器学习方法,发现在Ⅰ型黏多糖贮积症患者尿液中精氨酸等24种氨基酸、总糖胺聚糖、硫酸肝素和硫酸角质素的浓度显著升高,是可能的生物标志物[32]。Sharma等[26]基于主成分分析和支持向量机方法发现Ⅰ型戈谢病患者的骨小梁微结构指标与健康人群存在差异,使用该指标辅助诊断Ⅰ型戈谢病,准确率可达73%。
Kadali等[19]利用机器学习方法构建分类和回归树模型,用于黏多糖贮积症的鉴别诊断,准确率为96.3%,有助于直接帮助医生在诊断中决定进行哪些特定的酶活性测定。Andrade-Campos等[25]使用决策树算法等机器学习方法对西班牙戈谢病登记处中收集的数据进行分析,研究戈谢病晚期并发症的危险因素,发现既往脾切除、血清IgA水平、延迟治疗与骨骼并发症和严重骨病的发展具有相关性。
Wilson等[22]使用机器学习算法分别构建两种戈谢病预测模型(基于症状发生的年龄的算法和基于患病率的算法),在电子健康档案数据库中筛查具有高戈谢病风险的人群,诊断效率比临床诊断高10~20倍;Ehsani-Moghaddam等[28]使用朴素贝叶斯分类器在加拿大初级保健哨兵监测网络的电子医疗记录中诊断Ⅱ型黏多糖贮积症,准确度高达99%;Hilz等[31]建立法布雷病风险预测模型,临床应用的敏感度为80.4%,特异度为79.8%;Jefferies等[14]开发的法布雷病人工智能预测工具——OM1 Patient Finder,在临床验证计算出的受试者工作特征曲线下面积为0.82,具有较高的辅助诊断准确度。
2.2.4自然语言处理
Kuipers等[29]开发了一种名为Mutator的方法,利用文本挖掘和自然语言处理等技术,从大量文献中自动提取法布雷病相关的突变信息,有效提高更新突变数据库的效率。与现有的法布雷病突变数据库(人类基因突变数据库和Swiss-Prot)相比,Mutator从文献中额外提取了30%的突变信息。而且在Mutator所提取的100篇文章中只有6篇文章是假阳性(即提取的突变信息与法布雷病无关),假阳性率较低。
2.2.5计算机视觉
计算机视觉研究的目的是教会计算机去“看”,即从图像或多维数据中获取所需的信息,并辅助临床决策的人工智能系统。4项研究用面部分析算法Face2Gene识别戈谢病、法布雷病等LSD患者的性能,诊断准确率分别为62%[16]、89%[2,7]、57%[30]。两项研究[11-12]利用计算机辅助的半自动化软件计算法布雷病患者视网膜眼底图像上的血管迂曲度参数。与健康对照相比,法布雷病患者的角度和度量之和(sum of angle metric,SOAM)、角度距离乘积(product of angle distance,PAD)和三角形指数(triangular index,TI)显著增高[11],表明计算机辅助分析视网膜血管迂曲度可以有效识别法布雷病患者。一项研究[33]使用虹膜识别相机获取并计算黏多糖贮积症患者的角膜浑浊分数(corneal opacification measure,COM),发现COM与临床角膜浑浊分级之间存在强相关性,线性回归相关系数为8.31,调整后的R2为0.69,说明相比于临床上对黏多糖贮积症引起的角膜浑浊进行主观评价,运用图像分析算法评估虹膜相机图像具有有效性和可靠性。
3讨论
本文系统梳理了应用数智化技术开展LSD辅助诊断的现状。结果显示,数智化技术在LSD辅助诊断中的应用尚不完善,尤其是国内相关工作开展较少。针对已开展的研究进行剖析后发现,数智化技术在LSD辅助诊断领域的应用具有巨大的潜力和价值,能为提升诊断准确率、提高检测效率、促进精准诊疗发展提供重要支持。
当前开展应用的数智化技术主要包括大数据技术、机器学习技术、自然语言处理技术和计算机视觉技术4个类别。大数据存储及管理技术为多模态融合数据库的构建提供了技术支持。例如,amamutdb.no和Pompe Registry数据库不仅整合了多种基因变异和临床信息,还为疾病的整体理解与精准诊断提供了参考,能够在临床表型模糊或重叠的情况下,为医生提供决策支持。另外,利用数据分析及挖掘技术,研究者可从海量数据中准确提取用于致病机制探索和病因分析的有价值信息。例如,对Pompe Registry的扩展分析发现多个GAA基因的新突变位点。机器学习算法帮助研究者发现更多有诊断意义的生物标志物,提高疾病预测的敏感性和准确性。在信息提取和文献分析方面,自然语言处理技术可以在文献中自动提取疾病相关信息,实现数据的快速分类与整合。计算机视觉技术在面部分析与视网膜影像分析中的应用,使某些原本依靠医生主观判断的病症表现更加客观化、定量化,从而提高了诊断的规范性和准确性,简化了诊断方式,提高了诊断效率。
由于LSD的罕见性和疾病临床表型的多样性[2,3,7,14,22],传统诊断方法难以快速、准确地识别患者。相比之下,基于数智化技术的辅助诊断展现出更多优势。首先,具有较高的诊断效率和特异性。研究显示,将数据挖掘技术用于电子健康档案等大数据,能快速识别潜在的患者[20];基于机器学习算法的LSD筛查也呈现出较高的特异性[14,22,27,31]。其次,具有较强的精准性。一方面,数智化技术的应用帮助研究者和临床医生更好地理解基因突变与不同疾病表型之间的关系,为不同表型匹配相应的突变类型,助力疾病的精准诊断;另一方面,数智化技术还能帮助研究者筛选出更多的特异性生物标记,如法布雷病视网膜血管迂曲度[11]、黏多糖贮积症角膜浑浊度[33]等,为疾病的精准诊断提供支持。
尽管数智化技术在LSD辅助诊断领域的应用展现出广阔的前景,但也面临一些不足和挑战,具体如下:①对医疗健康大数据进行分析、挖掘与融合带来的患者知情同意和隐私保护方面的伦理问题[14,36];②罕见病有限的样本量和数智化技术对大样本的需求之间的矛盾[7,12,16,20,26];③基于罕见病“有限样本”获得的计算模型向“无限总体”推广应用产生的外推性受限和预测准确性下降的问题[19,28]。为此,充分借鉴数智化技术在常见病辅助诊疗中的应用经验,尝试提出可行的应对方案。首先,利用区块链技术或联邦学习技术实现在不同来源医疗健康数据融合与分析过程中对患者隐私的保护;第二,通过罕见病病例登记或利用电子病历等真实世界数据推动罕见病例数量的快速积累;第三,开展多中心病例登记或融合不同群体来源的医疗健康数据用于模型的构建与交叉验证,提高模型应用的普适性。此外,只将某一种数智化技术应用于疾病的辅助诊断非常有限,要充分发挥数智化技术“智”的特点,可以尝试将多种技术联合应用,例如,将机器学习与大数据分析挖掘技术相结合,在丰富的数据库中开展训练;或将计算机视觉、自然语言处理与机器学习结合,构建既能“看”也能“读”的数智化技术集成模型,全方位模仿医生的诊断过程,提高诊断的自动化程度和效率;同时,数智化辅助诊断技术应与医生的临床诊断紧密结合,而不是单纯替代,二者相辅相成,数智化技术帮助医生快速分析患者历史病历,从海量数据中挖掘有价值的信息,并进行诊断预测,医生则利用自己的临床经验,结合预测结果做出最终诊断,并为改进数智化技术提供建议;最后,数智化技术归根结底是基于“数”的,“数字化”是“数智化”的前提和基础。对多源数据的充分融合、深度挖掘和高效治理是构建计算最优模型的根本保障。
同时,在宏观层面也需要设计相应的机制,促进基础数据和技术的集成与整合。为此,笔者提出DI-HEALTH理论框架(Digital Intelligence-driven, multilevel hospital, accessible, affordable, collaboration, full-cycle health care),即以数智化技术为驱动,以信息平台为核心,整合多层级医院与多种类技术,为罕见病患者提供可及、可负担的全周期医疗照护服务。围绕LSD的辅助诊断,可充分利用已有的信息化平台,如中国国家罕见病注册系统(National Rare Diseases Registry System of China,NRDRS),实现在线患者信息采集、存储与管理,完成病例信息的积累;使用系统搭载的多种类数智化工具和模型处理、分析病例数据,生成疾病诊断预测结果,辅助医生进行诊断;以多终端移动设备为触手,有效连接基层社区和乡镇的卫生服务机构和患者,实现罕见病患者的远程诊疗与健康咨询,并通过5G技术,完成病例信息的即时采集与传输,不断扩充完善数据库。
本研究仍存在以下局限性:①受限于文献数据库的权限,本研究只针对常见的数据库进行检索,且未纳入中文和英文之外其他语种的文献;②纳入的文献均报道了数智化技术辅助LSD临床诊断的优势,可能存在发表偏倚;③现有关于数智化技术驱动LSD辅助诊断的研究较少,并且本文未关注该技术在不同亚类LSD患者中的使用效果,导致结论的外推性有限;④国内相关研究开展较少,因此,数智化技术驱动的辅助诊断是否适用于国人尚需进一步探索。
综上,数智化技术驱动的LSD辅助诊断已展现出巨大的价值和潜力,国内医疗和研究机构应结合国人特征,在DI-HEALTH理论框架的指导下,积极探索构建适用于中国LSD患者的数智化辅助诊断体系和技术,充分结合“数”与“智”各自的优势,将其在临床实践中进行推广和应用。
作者贡献:杜欣羽负责撰写初稿、筛选文献、提取文献信息;王胜锋负责文献筛选和提取信息核查、审阅和修订论文;谢静负责文献检索;郭健负责酝酿和设计实验、论文审阅和修改,以及文献筛选;张抒扬负责酝酿和设计实验,以及论文审阅和修改。
利益冲突:所有作者均声明不存在利益冲突。
参考文献
[1]Andrade-Campos M, Alfonso P, Irun P, et al. Diagnosis features of pediatric Gaucher disease patients in the era of enzymatic therapy, a national-base study from the Spanish Registry of Gaucher Disease[J]. Orphanet J Rare Dis, 2017,12(1):84.
[2]D'Souza A, Ryan E, Sidransky E. Facial features of lysosomal storage disorders[J]. Expert Rev Endocrinol Metab, 2022, 17(6):467-474.
[3]Ranieri M, Bedini G, Parati EA,et al. Fabry disease: recognition, diagnosis, and treatment of neurological features[J]. Curr Treat Options Neurol, 2016,18(7):33.
[4]Michaels-Igbokwe C, McInnes B, MacDonald KV, et al. (Un)standardized testing: the diagnostic odyssey of children with rare genetic disorders in Alberta, Canada[J]. Genet Med, 2021,23(2):272-279.
[5]Hartley T, Lemire G, Kernohan KD, et al. New diagnostic approaches for undiagnosed rare genetic diseases[J]. Annu Rev Genomics Hum Genet, 2020, 21:351-372.
[6]Curiati M, Aranda C, Kyosen S, et al. The challenge of diagnosis and indication for treatment in Fabry disease[J/OL]. J Inborn Errors Metab Screen, 2017, 5: 232640981668573.
[7]Daykin E, Fleischer N, Abdelwahab M, et al. Investigation of a dysmorphic facial phenotype in patients with Gaucher disease types 2 and 3[J]. Mol Genet Metab, 2021, 134(3):274-280.
[8]刘国斌,祁伯洋.县域城镇数智化与信息化融合发展研究[J].情报科学,2022,40(3):21-26.
[9]张建锋,肖利华,许诗军.数智化:数字政府、数字经济与数字社会大融合[M].北京:电子工业出版社,2022.
[10]王秉. 何为数智:数智概念的多重含义研究[J]. 情报杂志,2023,42(7):71-76.
[11]Sodi A, Guarducci M, Vauthier L, et al. Computer assisted evaluation of retinal vessels tortuosity in Fabry disease[J]. Acta Ophthalmol, 2013, 91(2):e113-e119.
[12]San Román I, Rodríguez ME, Caporossi O, et al. Computer assisted retinal vessel tortuosity evaluation in novel mutation Fabry disease: towards new prognostic markers[J]. Retina, 2017, 37(3):592-603.
[13]Siontis KC, Noseworthy PA, Attia ZI, et al. Artificial intelligence-enhanced electrocardiography in cardiovascular disease management[J]. Nat Rev Cardiol, 2021, 18(7):465-478.
[14]Jefferies JL, Spencer AK, Lau HA, et al. A new approach to identifying patients with elevated risk for Fabry disease using a machine learning algorithm[J]. Orphanet J Rare Dis, 2021,16(1):518.
[15]Tebani A, Mauhin W, Abily-Donval L, et al. A proteomics-based analysis reveals predictive biological patterns in Fabry Disease[J]. J Clin Med, 2020,9(5):1325.
[16]Pantel JT, Zhao M, Mensah MA, et al. Advances in computer-assisted syndrome recognition by the example of inborn errors of metabolism[J]. J Inherit Metab Dis, 2018, 41(3):533-539.
[17]Riise Stensland HM, Frantzen G, Kuokkanen E,et al. amamutdb.no: a relational database for MAN2B1 allelic variants that compiles genotypes, clinical phenotypes, and biochemical and structural data of mutant MAN2B1 in α-mannosidosis[J]. Hum Mutat, 2015, 36(6):581-586.
[18]Moreno-Barea FJ, Franco L, Elizondo D, et al. Application of data augmentation techniques towards metabolomics[J]. Comput Biol Med, 2022,148:105916.
[19]Kadali S, Naushad SM, Radha Rama Devi A, et al. Biochemical, machine learning and molecular approaches for the differential diagnosis of Mucopolysaccharidoses[J]. Mol Cell Biochem, 2019, 458(1-2):27-37.
[20]Moynihan D, Monaco S, Ting TW, et al. Cluster analysis and visualisation of electronic health records data to identify undiagnosed patients with rare genetic diseases[J]. Sci Rep, 2024, 14(1):5056.
[21]Ghasemi M, Rahgozar M, Kavousi K. Complex disease genes identification using a heterogeneous network embed-ding approach[J]. IEEE/ACM Trans Comput Biol Bioinform, 2023, 20(2):875-882.
[22]Wilson A, Chiorean A, Aguiar M, et al. Development of a rare disease algorithm to identify persons at risk of Gaucher disease using electronic health records in the United States[J]. Orphanet J Rare Dis, 2023, 18(1):280.
[23]Nio MY, In't Groen SLM, Bergsma AJ, et al. Extension of the Pompe mutation database by linking disease-associated variants to clinical severity[J]. Hum Mutat, 2019, 40(11):1954-1967.
[24]Reuser AJJ, van der Ploeg AT, Chien YH, et al. GAA variants and phenotypes among 1,079 patients with Pompe disease: data from the Pompe Registry[J]. Hum Mutat, 2019, 40(11):2146-2164.
[25]Andrade-Campos MM, de Frutos LL, Cebolla JJ, et al. Identification of risk features for complication in Gaucher's disease patients: a machine learning analysis of the Spanish registry of Gaucher disease[J]. Orphanet J Rare Dis, 2020,15(1):256.
[26]Sharma GB, Robertson DD, Laney DA, et al. Machine learning based analytics of micro-MRI trabecular bone microarchitecture and texture in type 1 Gaucher disease[J]. J Biomech, 2016, 49(9):1961-1968.
[27]Soares LDF, Villalba Silva GC, Kubaski F, et al. MPSBase: comprehensive repository of differentially expressed genes for mucopolysaccharidoses[J]. Mol Genet Metab, 2021, 133(4):372-377.
[28]Ehsani-Moghaddam B, Queenan JA, MacKenzie J, et al. Mucopolysaccharidosis type Ⅱ detection by Nave Bayes Classifier: an example of patient classification for a rare disease using electronic medical records from the Canadian Primary Care Sentinel Surveillance Network[J]. PLoS One, 2018,13(12):e0209018.
[29]Kuipers R, van den Bergh T, Joosten HJ, et al. Novel tools for extraction and validation of disease-related mutations applied to Fabry disease[J]. Hum Mutat, 2010, 31(9):1026-1032.
[30]Marwaha A, Chitayat D, Meyn MS, et al. The point-of-care use of a facial phenotyping tool in the genetics clinic: enhancing diagnosis and education with machine learning[J]. Am J Med Genet A, 2021,185(4):1151-1158.
[31]Hilz MJ, Lyn N, Marczykowski F, et al. Unveiling the untreated: development of a database algorithm to identify potential Fabry disease patients in Germany[J]. Orphanet J Rare Dis, 2024,19(1):259.
[32]Tebani A, Schmitz-Afonso I, Abily-Donval L, et al. Urinary metabolic phenotyping of mucopolysaccharidosis type I combining untargeted and targeted strategies with data modeling[J]. Clin Chim Acta, 2017,475:7-14.
[33]Aslam TM, Shakir S, Wong J, et al. Use of iris recognition camera technology for the quantification of corneal opacification in mucopolysaccharidoses[J]. Br J Ophthalmol, 2012, 96(12):1466-1468.
[34]Pode-Shakked B, Finezilber Y, Levi Y, et al. Shared facial phenotype of patients with mucolipidosis type Ⅳ: a clinical observation reaffirmed by next generation phenotyping[J]. Eur J Med Genet, 2020,63(7):103927.
[35]李新平. 大数据处理流程及存储与管理技术研究[J]. 电脑编程技巧与维护,2023,(3):97-100,133.
[36]Sobrido MJ, Bauer P, de Koning T, et al. Recommendations for patient screening in ultra-rare inherited metabolic diseases: what have we learned from Niemann-Pick disease type C?[J]. Orphanet J Rare Dis, 2019, 14(1):20.