


205、血管性血友病3型
罕见病诊疗指南(2025年版)
概述 血管性血友病(von Willebrand disease, VWD)是由于血管性血友病因子(von Willebrand factor, VWF)质或量的异常导致的一类遗传性出血性疾病。VWF在体内主要参与血小板黏附及保护凝血因子Ⅷ,在生理性止血过程中发挥重要作用。根据VWF异常特征,VWD主要分为三种类型:1型为VWF量的部分减少;2型为VWF质的缺陷,3型为VWF量的显著减少或缺如。临床上3型VWD较为少见,但出血程度严重,常自幼发病,除皮肤黏膜出血外,常发生内脏、肌肉、关节出血,严重内脏出血或颅内出血可危及生命。
病因和流行病学
血管性血友病(VWD)主要是由于血管性血友病因子(VWF)基因突变所致,造成VWF合成和分泌减少或异常,引起血浆VWF数量减少或质量异常。VWD符合常染色体显性或隐性遗传规律,而3型VWD为常染色体隐性遗传规律,VWF等位基因发生纯合突变或双重杂合突变,VWF合成缺如。人群中VWD发病率报道数据不一,如果按照血浆VWF水平降低来诊断VWD,预计发病率为1%。但从有明显出血症状同时伴VWF水平降低判断,VWD的发病率为1/1000。3型VWD较为罕见,占全部VWD病例的1%~5%,其发病率为(0.1~5.3)/100万。
临床表现
1.自幼发病,以皮肤、黏膜出血为主,表现为皮肤瘀点瘀斑、鼻出血和牙龈出血,严重者可伴有不同程度的缺铁性贫血;女性月经出血增多最为突出,严重者常伴有反复黄体破裂出血等急腹症。3型VWD患者也可发生关节、肌肉出血,消化道出血,颅内出血等。
2.多为自发性出血或外伤、围手术期出血过多。
3.部分患者有出血家族史,有家族史者符合常染色体隐性遗传规律。
由于VWF水平受种族、血型、年龄、炎症、妊娠等多种因素影响,患者的出血表现存在个体差异,对于初诊的出血患者,推荐使用出血评分工具(blood assessment tools,BATs)进行评估,根据评估结果决定是否需要行进一步的实验室检查。
辅助检查
1.出血筛选试验
(1)全血细胞计数,3型VWD患者血小板计数正常,可有不同程度的小细胞低色素性贫血。
(2)APTT/ PT,3型VWD患者APTT常延长,且可以被正常血浆纠正;PT正常。
(3)血浆纤维蛋白原测定,3型VWD患者纤维蛋白原水平正常。
筛选试验主要目的是排除其他原因所致的出血,3型VWD患者筛选检查结果仅有APTT延长且可被正常血浆纠正。
2.VWD诊断试验
(1)血浆VWF抗原测定(VWF:Ag),3型VWD患者VWF:Ag<3%。
(2)血浆VWF血小板结合活性测定:可采用VWF与野生型GPⅠb结合测定(VWF:GPⅠbR)、VWF与GPⅠb突变体结合测定(VWF:GPⅠbM)、VWF瑞斯托霉素辅因子活性(VWF:RCo)三种方法检测。VWF:GPⅠbR和VWF:GPⅠbM检测方法重复性好,受影响因素小,检测下限更低。3型VWD患者血浆VWF血小板结合活性<3%。
(3)血浆凝血因子Ⅷ活性测定(FⅧ:C),3型VWD患者血浆因子Ⅷ凝血活性显著降低,常<10%。
3.VWD分型诊断试验
(1)血浆VWF多聚体分析:3型VWD患者VWF多聚体缺如。
(2)瑞斯托霉素诱导的血小板聚集(RIPA),3型VWD患者RIPA显著降低。
(3)血浆VWF胶原结合试验(VWF:CB),3型VWD血浆VWF:CB显著降低。
(4)血浆VWF因子Ⅷ结合活性(VWF:FⅧB),3型VWD血浆VWF:FⅧB显著降低。
(5)VWF前导肽(VWF propeptide, VWFpp)测定,3型VWD患者VWFpp水平和VWF:Ag水平一样,显著下降。
(6)VWF基因测序:随着二代基因测序技术的发展,VWF基因测序在VWD的诊断中发挥了重要作用,3型VWD患者中VWF突变检出率90%以上。
诊断
1.有家族史者,符合常染色体隐性遗传规律。
2.有自发性出血或外伤、围手术期出血增多史,并符合VWD临床表现特征。
3.血浆VWF水平(包括VWF:Ag和VWF血小板结合活性)<3%。
4.排除血友病A、获得性血友病A、获得性von Willebrand综合征(acquired von Willebrand syndrome, AVWS)、血小板型VWD、遗传性血小板病等。
鉴别诊断
1.血友病A 是遗传性凝血因子Ⅷ缺乏所导致的一种X连锁隐性遗传病。女性携带、男性发病。临床上关节、肌肉出血为其主要特点,VWF水平正常。
2.获得性血友病A 是由于体内产生抗凝血因子Ⅷ自身抗体导致凝血因子Ⅷ水平下降的获得性出血性疾病。患者的APTT延长且不能被正常混合血浆所纠正,凝血因子Ⅷ抑制物阳性,VWF水平正常。
3.获得性von Willebrand综合征(AVWS) 是一种少见的获得性出血性疾病。部分患者可继发于甲状腺功能减退、淋巴增殖性疾病、自身免疫性疾病或心血管疾病等。由于体内产生抗VWF自身抗体导致VWF功能受损或VWF清除增快。患者既往无类似出血史和家族性出血性疾病史,有基础疾病的患者随着原发病控制,VWF水平和出血症状可获得改善;通过血浆混合试验或ELlSA方法可检测vWF的自身抗体
4.血小板型VWD 系血小板膜糖蛋白GPIbα突变导致与VWF结合能力增强,血浆大分子VWF多聚体减少。患者常出现不同程度的血小板减少,血小板与正常人血浆混合后,能被低浓度瑞斯托霉素诱导血小板聚集,检测到血小板膜糖蛋白GPIbα基因突变有助于诊断。
5.遗传性血小板病 是一组先天性血小板数量或功能异常的疾病,临床上患者可有不同程度的出血表现。但患者VWF水平和功能通常正常。
治疗
3型VWD患者主要治疗目的是预防和控制出血。目前,3型VWD患者主要治疗方法是替代治疗,即补充不足的VWF恢复正常止血功能。3型VWD患者出血发作时给予的替代治疗是以控制出血为目的;围手术期进行替代治疗以预防出血为目的;对出血表现严重或有关节肌肉出血、颅内出血史者应进行长期规律性预防治疗。
1.替代治疗选用血源性/重组VWF制剂或血源性含VWF的FⅧ浓缩制剂;如条件限制也可使用冷沉淀物或新鲜血浆,但存在输血相关疾病传播风险。使用剂量依出血部位和出血程度而定。剂量标定以制剂的VWF:RCo为准,每公斤体重输注1 U的VWF:RCo平均使血浆VWF:RCo提高2 U/dL 推荐剂量:严重出血或大型手术者,首次或术前用量40~60 U/kg,维持量20~40 U/kg,每12~24h一次,持续3~14天;中度出血或小型手术者,首次或术前用量30~60 U/kg,维持量20~40 U/kg,每12~24h一次,持续1~5天;轻度出血可单次使用,20~30 U/kg。
预防治疗剂量和频率个体差异较大,多为20~40 U/kg/次,每周2次。国外相关文献已经证实预防治疗的长期效果。
有文献报道对3型VWD患者探索性使用非因子制剂进行预防出血,如艾美赛珠单抗,以及即将上市的凝血再平衡药物(ATIII抑制剂和TFPI抑制剂)等。
2.其他治疗
(1)抗纤溶药物①氨甲环酸:每次25mg/kg,口服,一日三次,或每次15mg/kg,静脉滴注,8小时一次。②6-氨基己酸:首剂4~5g,静脉滴注,后每小时1g至出血控制,24小时总量不超过24g。抗纤溶药物偶有血栓形成危险,血尿者禁用。牙龈出血时可局部使用,也可作为VWD患者出血或手术时的辅助治疗。
(2)局部使用凝血酶或纤维蛋白凝胶对皮肤黏膜出血治疗有辅助作用。
(3)替代治疗止血效果不佳者可考虑使用重组活化凝血因子Ⅶ。
3.女性3型VWD患者的治疗
(1)伴月经增多的3型VWD患者 首先需排除其他与月经增多相关的妇科疾病。对于没有生育要求的患者,采用性激素治疗(复合激素避孕药或左炔诺孕酮释放宫内缓释系统)或者氨甲环酸。对于有生育需求的患者,首选氨甲环酸。如单药治疗效果欠佳,可联合使用替代治疗。子宫内膜切除术或子宫全切术仅适用于常规治疗无效的VWD患者。反复月经过多患者注意评估缺铁和贫血状态,予以铁剂治疗。
(2)出血性卵巢囊肿女性3型VWD患者可发生出血性卵巢囊肿或黄体破裂出血,引起急腹症。治疗方法包括VWF替代治疗、抗纤溶药物等,对重症患者须急症手术治疗。术后性激素治疗可预防复发。
(3)妊娠及分娩VWD妇女可正常妊娠,但出血、流产的发生率增高,多发生于妊娠期前3个月。分娩时如采用神经阻滞麻醉,VWF水平需维持在50%~150%,直至麻醉结束后6小时。分娩后VWD患者还存在产后出血风险,需密切观察,必要时口服氨甲环酸,或持续予以替代治疗至恶露期结束。此外,需评估新生儿罹患VWD的风险,出生后可采集脐带血检查VWF水平。
4. 3型VWD患者其他特殊情况的治疗
(1)围手术期治疗VWD患者进行大手术时,FⅧ:C和VWF水平保持在50%以上并维持至术后3天,后根据病情减量使用。进行小手术或有创操作时,推荐用因子浓缩物提升VWF水平大于50%,同时联合氨甲环酸治疗。大手术是指需要进入胸腹盆腔的、可能出现严重出血、涉及关节的手术,还包括拔除第三磨牙以及危及患者生命的手术(如颅脑手术、喉部手术等)。而简单拔牙、门诊手术和那些大手术之外的手术,则定义为小手术。
(2)3型VWD患者合并心血管疾病或血栓性疾病需要抗血小板或抗凝治疗时,应在充分评估患者的血栓和出血风险后酌情在预防治疗的基础上给予必要的抗血小板或抗凝治疗。
综上所述,3型VWD患者通常有明显的出血症状,这类患者面临手术、妊娠、分娩或心脑血管等疾病时,建议患者至有经验的VWD综合管理中心,由血液专科、手术科室、妇产科、麻醉科或其他涉及科室的医生组成多学科诊疗团队共同权衡利弊,制定合适的治疗方案。
诊疗流程(图84-1)
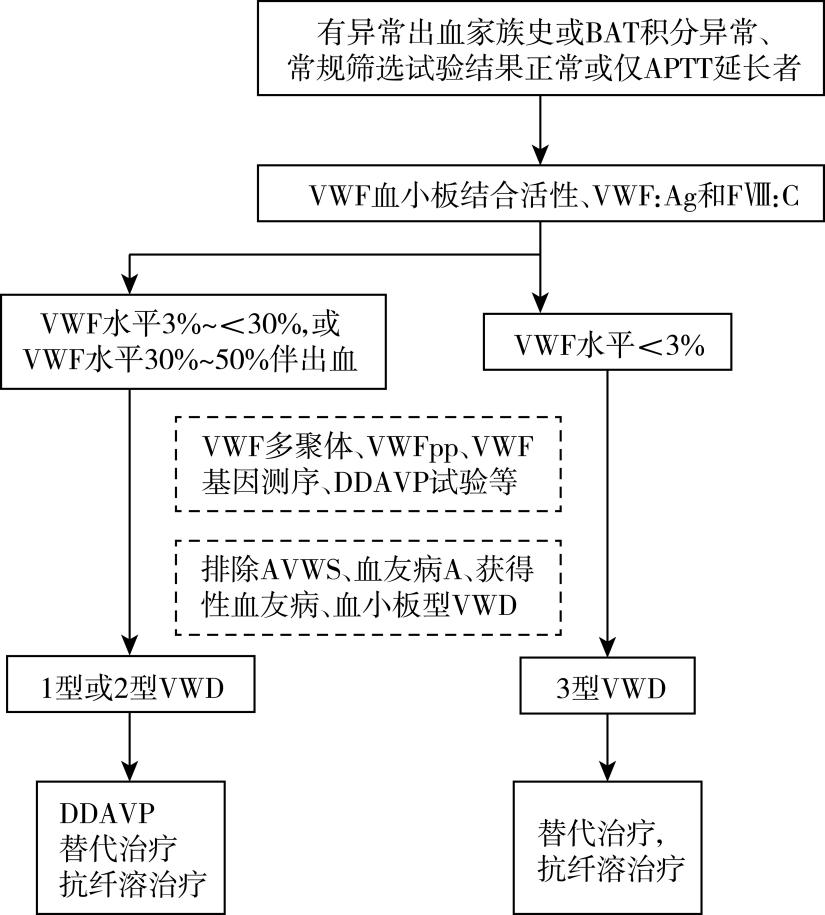
图84-1 VWD诊疗流程图
参考文献
[1] 中华医学会血液学分会血栓与止血学组.血管性血友病诊断与治疗中国指南(2022年版).中华血液学杂志,2022, 43(1): 1-6.
[2] FOGARTY H, DOHERTY D, O'DONNell JS. New developments in von Willebrand disease. Br J Haematol, 2020, 191(3): 329-339.
[3] SHARMA R, HABERICHTER SL. New advances in the diagnosis of von Willebrand disease. Hematology Am Soc Hematol Educ Program,2019,2019(1):596-600.
[4] PAGLIARI MT, BUDDE U, BARONCIANI L,et al.von Willebrand factor neutralizing and non-neutralizing alloantibodies in 213 subjects with type 3 von Willebrand disease enrolled in 3WINTERS-IPS.J ThrombHaemost, 2023,21(4):787-799.
[5] ELBATARNY M, MOLLAH S, GRABELL J, et al. Normal range of bleeding scores for the ISTH-BAT: adult and pediatric data from the merging project. Haemophilia, 2014, 20(6): 831-835.
[6] RODEGHIERO F, TOSETTO A, ABSHIRE T, et al. ISTH/SSC bleeding assessment tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J ThrombHaemost, 2010, 8(9):2063-2065.
[7] CASTAMAN G. How I treat von Willebrand disease. Thromb Res, 2020, 196: 618-625.
[8] JAMES PD, CONNELL NT, AMEER B, et al. ASH ISTH NHF WFH 2021 guidelines on the diagnosis of von Willebrand disease. Blood Adv, 2021, 5(1): 280-300.
[9] JAMES AH, EIKENBOOM J, FEDERICI AB. State of the art: von Willebrand disease. Haemophilia, 2016, 22(Suppl 5): 54-59.
[10] LEEBEEK FWG, ATIQ F. How I manage severe von Willebrand disease. Br J Haematol, 2019, 187(4): 418-430.
[11] GIANCARLO C, PAULA DJ. Pregnancy and delivery in women with von Willebrand disease. Eur J Haematol, 2019, 103(2): 73-79.
[12] ZULFIKAR B, KOC B, AK G, et al. Surgery in patients with von Willebrand disease. Blood Coagul Fibrinolysis, 2016,27(7): 812-816.
[13] CONNELL NT, FLOOD VH, BRIGNARDELLO-PETERSEN R, et al. ASH ISTH NHF WFH 2021 guidelines on the management of von Willebrand disease. Blood Adv, 2021, 5(1): 301-325.
PIEL-JULIAN ML, THIERCELIN-LEGRAND MF, MOULIS G, et al. Antithrombotic therapy management in patients with inherited bleeding disorders and coronary artery disease: a single-centre experience. Haemophilia, 2020, 26(2):e34-e37.