


199、地中海贫血(重型)
罕见病诊疗指南(2025年版)
概述 地中海贫血(thalassemia)是人类最常见的一种异质性隐性遗传的单基因疾病,由于珠蛋白生成不足或缺失导致不同程度的贫血,包括α地中海贫血和β地中海贫血,在地中海人群、中东、印度次大陆和缅甸都有较高的基因频率,我国长江以南10个省份也是地中海贫血的高发区。地中海贫血(重型)(Thalassemia major)为重型珠蛋白生成障碍性贫血,包括重型α地中海贫血和重型β地中海贫血。重型α-地中海贫血通常在宫内死亡或者出生后不久死亡。重型β-地中海贫血常于婴儿期发病,呈慢性进行性溶血性贫血,严重威胁患儿生存质量甚至生命。如无特别说明,本节中的地中海贫血(重型)专指重型β-地中海贫血。
病因和流行病学
1.α地中海贫血(α地贫,α thalassemia)由位于染色体16p13.3区域内的HBA1和HBA2基因突变引起,突变又可分为缺失型和非缺失型,其中缺失型突变占大多数。根据缺失的α基因数目,可分为α+地贫(缺失1个α基因,-α/)和α0 地贫(缺失2个α基因,--/)。非缺失型突变(αTα或ααT)指HBA1或HBA2基因发生点突变或若干碱基的缺失,通常被定义为α+地贫。血红蛋白H病是由4个α珠蛋白基因中的3个突变引起的,而血红蛋白Barts(Hb Bart水肿胎综合征)的所有4个α珠蛋白基因均发生突变。迄今为止,在中国人群中已发现了104种α珠蛋白基因突变。
2.β地中海贫血(β地贫,β thalassemia)由位于染色体11p15.4区域内的HBB基因的点突变或小片段缺失引起,染色体上的2个等位基因都有致病突变的个体称为纯合子;同源染色体上只有1个致病突变的个体称为杂合子;2个等位基因的致病突变不同的个体称为复合杂合子。重型β地贫的基因型为纯合子或复合杂合子状态。若双亲均为β地贫杂合子,其子女获得重型β地贫的概率为25%,杂合子概率为50%,余25%为正常。
3.溶血和无效造血是地中海贫血的主要病理生理学机制。α/β珠蛋白肽链合成的不平衡引起过多的β珠蛋白或α珠蛋白在红细胞前体中积聚并沉淀,使红细胞受损,导致慢性溶血性贫血。尽管造血旺盛,重型地中海贫血患者多数红系祖细胞均在骨髓中死亡,导致无效造血;代偿性骨髓造血增加,造成骨骼变形,形成地贫面容,引起脾脏增大。无效造血还可能造成机体铁过载,引起脏器功能损伤、生长发育迟缓,严重者可能危及生命。大约5%的世界人口携带α珠蛋白基因的变异。在我国,α地贫基因携带率最高的为广东(12.70%)、广西(19.11%)和海南(45.04%)三省份;香港、台湾、福建、江西、云南、贵州等省区也是携带率较高的地区。β地贫基因人群突变携带率为2%~30%,我国β地贫基因携带率最高的三个省份为广西(6.66%)、海南(5.11%)和贵州(4.63%),广东、云南、香港、湖南、江西等地区亦较高。据不完全统计,我国除香港、澳门、台湾地区以外的大陆范围内现有地贫基因携带者约为3000万人,中间型、重型地贫患者约30万人,其中重型β地贫患者1.5万人,纳入第二批罕见病目录。
临床表现
1.α地贫的临床表现变异度较大,病情的严重程度与α珠蛋白链减少的程度直接相关。Hb Bart’s水肿胎综合征又称重型α地贫,为致死性疾病。受累胎儿由于严重贫血和缺氧,常在妊娠23~38周在宫内或分娩后半小时内死亡。胎儿全身水肿、黄疸、肝脾明显肿大、发育不良。怀有此类胎儿的孕妇可能发生先兆子痫、早产和异常出血;由于巨大胎盘,分娩时可引发严重的并发症。
2.重型β地贫患儿出生时无症状,3~12月龄开始发病,呈慢性进行性贫血,面色苍白,肝脾逐渐肿大,体格发育逐渐落后,常有轻度黄疸。长期重度贫血使骨髓代偿性增生导致骨骼变大、髓腔增宽:1岁后颅骨改变明显,表现为头颅变大、额部隆起、颧高、鼻梁塌陷,两眼距增宽,为地贫特殊面容。症状体征随年龄增长而日益明显。患儿因长期贫血致免疫功能低下,常并发支气管炎或肺炎。当合并含铁血黄素沉着症时,因过多的铁沉着于心肌和肝、胰腺、脑垂体等其他器官,而引起该器官功能受损的相应症状,包括合并凝血功能障碍、糖代谢异常、生长发育迟缓、骨质疏松等;其中最严重的是心力衰竭,它是贫血和铁沉积造成心肌损害的结果,是导致患儿死亡的主要原因之一。本病如不治疗,患儿多于5岁前死亡。
辅助检查
1.血液学改变 ①外周血血红蛋白(Hemoglobin, Hb)< 60g/L,呈小细胞低色素性贫血,红细胞平均容积(Mean corpuscular volume, MCV)< 80fL、红细胞平均血红蛋白(Mean corpuscular hemoglobin, MCH)< 27pg、红细胞平均血红蛋白浓度(Mean corpuscular hemoglobin concentration, MCHC)< 320g/L。红细胞形态不一,大小不等,中央淡染区扩大,出现靶形红细胞和红细胞碎片,网织红细胞增高。部分患儿由于骨髓造血代偿可致血小板增高。脾功能亢进时,白细胞和血小板减少。②骨髓象呈红细胞系统增生明显活跃,以中、晚幼红细胞占多数,成熟红细胞改变与外周血相同。③红细胞渗透脆性明显降低。
2.血红蛋白电泳 α地贫患者血红蛋白组成分析将显示血红蛋白A2(HbA2)下降,在某些情况下可检出Hb H和Hb Bart’s。当α地贫合并β地贫时,HbA2可在正常范围。重型β地中海贫血首诊时血红蛋白电泳显示胎儿血红蛋白(Fetal hemoglobin, HbF)显著增高,一般达30%~90%,是诊断重型β地贫的重要依据。HbF不增高应排除近期输血的影响,可在输血后3个月左右复查。血红蛋白电泳β-地贫携带者血红蛋白A2(HbA2)含量升高,重型β-地贫HbA2通常不升高。
3.区域及家系调查 区域调查显示患儿来自地贫高发区域。患儿父母亲外周血常规呈小细胞低色素性贫血,血红蛋白组成异常;基因检测证实为地贫基因携带者。
4.基因诊断 可采用等位基因特异性寡核苷酸探针点杂交(allele-specific oligonucleotide polymerase chain reaction, PCR-ASO)、反向点杂交(Reverse dot blot, RDB)、跨越断裂点PCR(Gap-PCR)、基于实时荧光PCR的探针熔解曲线分析(PCR melting curve analysis,PMCA)和DNA测序等方法检测地贫基因缺陷的类型。
诊断
根据重型地贫临床表现、小细胞低色素性贫血、异常血红蛋白组成及家系调查此4点可作出地中海贫血(重型)的临床诊断。有条件者均应进行基因诊断,基因型为纯合子或复合杂合子为确诊本病的指标。
鉴别诊断
常见的需要与地中海贫血(重型)鉴别诊断的疾病包括:
1.缺铁性贫血 外周血常规示小细胞低色素性贫血,但缺铁性贫血多有缺铁病因,无溶血证据,有红细胞游离原卟啉(Free erythrocyte protoporphyrin, FEP)升高、血清铁及铁蛋白降低、铁剂治疗有良好反应等特点。
2.红细胞葡萄糖-6-磷酸脱氢酶(Glucose-6-phosphate dehydrogenase, G-6-PD)缺乏所致先天性非球形细胞性溶血性贫血(Congenital non-spherocytic hemolytic anemia, CNSHA)。溶血严重者与地中海贫血(重型)临床表现相似。但前者在感染及服用氧化性药物后可加重贫血,红细胞Heinz小体阳性,HbF含量正常。
3.遗传性球形红细胞增多症 外周血涂片示红细胞呈小球形,红细胞渗透脆性及孵育渗透脆性增加可鉴别。
4.慢性自身免疫性贫血 由于机体出现抗自身红细胞膜的免疫抗体,使红细胞破坏所致溶血,Coombs试验阳性可鉴别。
5.幼年型粒单核细胞白血病 患儿有脾大表现,且血红蛋白电泳示HbF升高,但外周血和骨髓可出现幼稚白血病细胞,伴有染色体异常和相关基因突变可鉴别。
治疗
目前针对地中海贫血(重型)的治疗方法有以下3种:①规范性输血和祛铁治疗(需终生维持);②造血干细胞移植(可治愈疾病但依赖配型和患者的身体状况);③基因治疗及新药治疗(已取得重要进展,但成为临床常规尚需时日)。
1.输血治疗 其目的在于使患者的Hb浓度接近于正常水平。研究表明维持Hb> 90g/L才能基本保证患儿生长发育和日常活动,抑制骨髓及髓外造血,并将铁负荷控制在最低限度。推荐的方案为:①在Hb< 90g/L时启动输血计划;②每2~5周输血1次,每次输红细胞0.5~1U/10kg,时间为2~4小时;③通过输血将Hb维持在90~140g/L;④重度贫血患者每次输注的红细胞量宜少,速度宜慢,可少量、多次。铁过载是影响重型β地贫患者生存质量的重要因素。当输血≥ 10次、血清铁蛋白> 1000μg/L或肝铁浓度> 7mg/g干重时,应启动祛铁治疗,并每月监测血清铁蛋白,每年检测肝铁浓度。当血清铁蛋白< 1000μg/L或肝铁浓度< 7mg/g干重时,可暂停使用铁螯合剂。目前临床使用的铁螯合剂主要包括去铁胺、去铁酮和地拉罗司等。
2.造血干细胞移植 是目前治愈地中海贫血(重型)的主要方法。根据干细胞的来源,又可分为骨髓移植、外周血干细胞移植和脐血干细胞移植,其要点如下:①移植前患者风险评估:国际上通常采用佩萨罗标准。移植前患者的3种风险因素评分为肝大、肝纤维化以及铁螯合剂应用史。有条件的患者应尽早(2~7岁)接受移植;②供体选择:以人类白细胞抗原(human leukocyte antigen,HLA)配型选择供体:选择顺序为HLA全相合同胞供者>非血缘相关的HLA全相合供者>半相合供者;③移植预处理方案:经典的清髓方案为白消安及环磷酰胺。为减少排斥率,在预处理方案中可酌情加用抗胸腺球蛋白和氟达拉宾(Fludarabine)。
3.基因治疗 是基于基因修正的自体造血干细胞移植。目前β地贫的基因治疗可分为基因替代疗法和基因编辑疗法两种。已有临床前和临床研究证明基因治疗的可行性和有效性,然而,基因治疗临床经验有限,需要更多的临床数据和大规模试验来证明基因治疗是一种安全和治愈性的治疗方法。
4.罗特西普(luspatercept) 是一种晚期红细胞成熟剂,可促进地贫患者骨髓内幼红细胞向晚期红细胞分化成熟,适用于治疗需要定期输注红细胞且红细胞输注≤ 15单位/24周的≥ 18岁β地贫成人患者;推荐起始剂量为1.0 mg/kg,皮下注射,每3周1次;以1.0 mg/kg起始剂量至少连续给药2次(6周)后未达到红细胞输注负荷降低,则应将剂量增加至1.25 mg/kg。最大治疗剂量不应超过每3周1.25 mg/kg。目前,罗特西普对儿童患者的疗效和安全性的相关研究正在进行中。
5.脾切除术 为治疗地中海贫血(重型)患儿的姑息手段,脾切除的指征为:①依赖输血量明显增多,如维持Hb 90~105g/L,每年红细胞输注量> 200ml/kg者,且经规则祛铁治疗而铁负荷仍增加。②脾功能亢进者,患儿出现红细胞破坏增加,持续的白细胞减少或血小板减少,临床上出现反复感染或出血。③脾脏增大并有伴随症状者,如患儿出现明显左上腹疼痛或易饱感,巨脾引起压迫及脾破裂等可能。符合以上指征之一可行脾切除术,建议行脾切除术时患儿年龄≥ 5岁,5岁以下进行脾切除会增加严重败血症发生的风险。
疾病预防
开展人群普查和遗传咨询、做好婚前指导,对预防本病有重要意义。Hb Bart's水肿胎属于胎儿期致死性疾病,孕育这类胎儿的孕妇因巨大胎盘有发生大出血等严重并发症的风险,因此推荐对这类胎儿进行产前诊断,并在确诊后与其双亲沟通,在知情同意和符合伦理的前提下,选择性终止妊娠。在我国地贫高发地区推广专项婚前检查,如夫妇双方均为β地贫基因携带者,可于妊娠第8~12周吸取绒毛,或妊娠第16~20周抽取羊水分离脱落细胞,或妊娠第20周后抽取脐带血,通过基因诊断方法检测胎儿β地贫缺陷基因。在妊娠早期对重型β地贫胎儿做出诊断并及时由夫妇选择终止妊娠,以避免重型β地贫患儿出生,是目前预防本病行之有效的方法。
诊疗流程(图78-1)
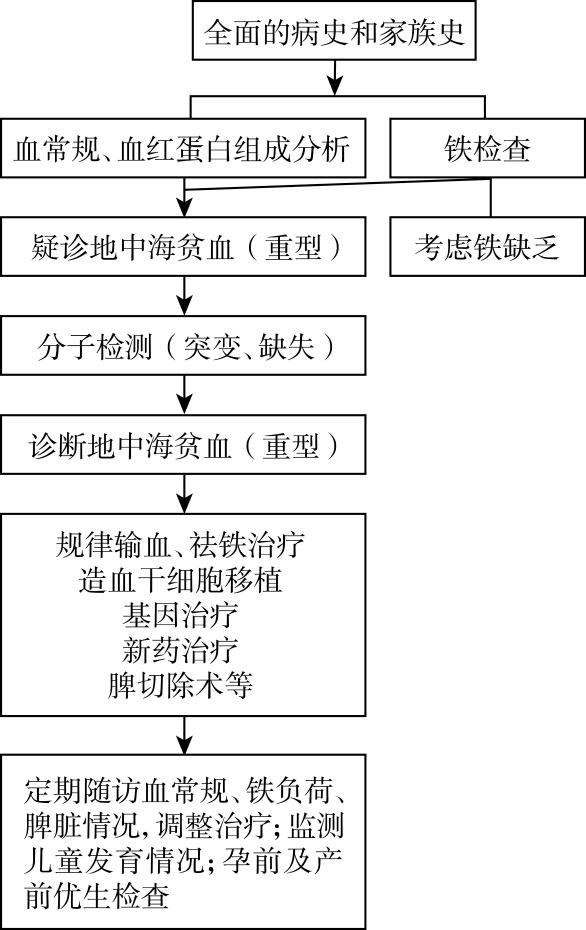
图78-1 地中海贫血(重型)诊疗流程
患者管理
地中海贫血是一类遗传性疾病,推荐建立患者管理平台,多学科共同照护,执行地贫患者信息化建设体系管理模式,是医疗护理服务的发起点和落脚点。
1.推荐地贫专科诊治医生 地贫专科诊治医生执行网络平台信息化建设体系,规范化诊疗管理模式,体现高质量诊疗水平。网络平台信息化建设体系需完善患者基本信息、建立个体化信息档案并做好各项诊疗记录。强化规范化输血和祛铁治疗的重要性,鼓励患者家庭共同参与;通过平台规范建立患者各项指标数据库,给予全程化精准照护,包括:
定期监测血红蛋白水平。每次输血前后须监测患者的血红蛋白等指标,以评估患者的贫血程度和可能的并发症,采取措施以改善贫血状态。
规范化输注血制品管理。地贫患儿的血红蛋白水平要尽量维持在90g/L以上,规范化输血可保障患儿的生长发育水平,使患儿的生长发育水平能够接近同龄健康儿童。患儿贫血状态改善,可提高其生活质量。
积极有效祛铁治疗。患儿血制品输注过多,铁沉积会加重心脏、肝脏等重要脏器的负担,可能会出现不良并发症。定期监测血液中铁蛋白的含量,推荐通过磁共振成像(MRI)了解心铁、肝铁沉积程度,结合患者临床信息的具体情况,给予有效祛铁治疗。
阶段监测生长发育。有报道指出约1/3重型β地贫患者在10岁时已经发生内分泌并发症,在2~9岁患者群体中30.5%存在内分泌疾病。推荐患儿10岁前开始监测生长发育及内分泌系统水平。建议体格检查与功能状态评估项目:身高、体重、头围、胸围、眼距、骨龄、维生素D;推荐生长发育检查项目:甲状腺功能、生长激素、性激素(男性12~18岁,女性9~18岁)监测等。
2.推荐血液病基因治疗专科护士 设立血液病基因治疗专科护士,完善地贫患者居家-入院-随访的全程化精准护理体系。建立地贫患者信息化管理平台,做好信息收集、横断面调研、健康科普等数据化支持。
3.建议提高患者就医黏附性 患者黏附性是治疗的关键点,为患者提供个性化、文化性的专病管理建议和服务,增强患者对专业团队的黏附性和依从性。医疗服务做“加法”、就医流程做“减法”、诊治能力做“乘法”、患者负担做“除法”,以帮助患者在医疗服务过程中感受到舒适化、智慧化和数字化,实现地贫患者家庭的真正的“脱贫”。
4.鼓励地贫家庭参与医疗安全 地贫患者的生理及心理负担极其严峻,导致患儿家庭的经济负担和照护负担增加,生活质量很差。鼓励患者和家庭能够积极参与医疗安全,赋予家庭权利,让他们积极参与到治疗照护中;诊疗过程中向患者和家属提供有效安全信息,保障各项医疗行为的顺利进行;为患者和家属提供疾病相关预防及促进健康知识,科普基因治疗,为患者和家属树立战胜疾病的信心。
参考文献
[1] 中华医学会儿科学分会血液学组,《中华儿科杂志》编辑委员会.重型β地中海贫血的诊断和治疗指南(2017年版).中华儿科杂志,2018,56 (10):724-729.
[2] 中华医学会血液学分会红细胞疾病(贫血)学组.中国输血依赖型β地中海贫血诊断与治疗指南(2022年版).中华血液学杂志, 2022,43(11):889-896.
[3] 中华医学会医学遗传学分会遗传病临床实践指南撰写组,商璇,吴学东,等. β-地中海贫血的临床实践指南.中华医学遗传学杂志,2020,37(3):243-251.
[4] 中华医学会医学遗传学分会遗传病临床实践指南撰写组,商璇,张新华,等. α-地中海贫血的临床实践指南.中华医学遗传学杂志,2020,37(3):235-242.
[5] 程涛.中国重型β地中海贫血患者疾病负担及诊疗现况横断面调查研究报告.北京:中华医学电子音像出版社,2023.
[6] WANG WD, HU F, ZHOU DH, et al. Thalassaemia in China. Blood Rev, 2023,60:101074.
[7] PEPE A, PISTOIA L, GAMBERINI MR, et al. National networking in rare diseases and reduction of cardiac burden in thalassemia major. Eur Heart J, 2022,43(26):2482-2492.
[8] CAPPELLINI MD, VIPRAKASIT V, TAHER AT,et al. A phase 3 trial of luspatercept in patients with transfusion-dependent β-thalassemia. N Engl J Med, 2020,382(13):1219-1231.
[9] THOMPSON AA, WALTERS MC, KWIATKOWSKI J, et al. Gene Therapy in Patients with Transfusion-Dependent β-Thalassemia. N Engl J Med, 2018,378(16):1479-1493.
[10] FRANGOUL H, ALTSHULER D, CAPPELLINI MD,et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N Engl J Med, 2021,384(3):252-260.