


187、原发性免疫缺陷病
罕见病诊疗指南(2025年版)
概述 原发性免疫缺陷病(primary immunodeficiency disease,PID)是一类由单基因突变导致免疫细胞或分子数量和/或功能异常导致的一类疾病,引起机体易患感染、过敏、自身免疫、自身炎症及肿瘤等疾病。
1970年世界卫生组织(World Health Organization,WHO)首次对PID进行了分类和定义,国际免疫学会联合会(International Union of Immunology Societies,IUIS)专家组每2~3年对PID进行分类更新。2017年IUIS专家组将PID更名为免疫出生缺陷(inborn errors of immunity,IEI),以避免对PID出现片面的认知和理解。但PID仍在临床上被广泛沿用。
目前,国际免疫学会专家委员会将PID分为10大类:联合免疫缺陷病、伴典型表现的联合免疫缺陷综合征、抗体缺乏为主的免疫缺陷病、免疫失调性疾病、吞噬细胞缺陷、固有免疫缺陷、自身炎症性疾病、补体缺陷、单基因骨髓衰竭综合征及拟表型。需注意的是,某些疾病归类比较特殊,可归入不同分类。特别是随着近代免疫学技术的突飞猛进,人们对PID的认识越来越精准。人们越来越意识到,PID临床表现多样,异质性非常强,常常需个体化分析归类。例如,CD40配体(CD40L)基因突变所致的高IgM综合征被归入联合免疫缺陷病和抗体缺乏为主的免疫缺陷病两类。因为该病大部分患儿具有联合免疫缺陷病临床表现,但亦有少数可在IVIG替代治疗后存活至成人期,并没有明显的机会感染。为了更充分反映这些PID的临床和免疫学特点,因此将其归入不同分类,方便治疗和长期随访管理。
近年来,随着基础免疫学的快速发展和高通量测序技术在临床的广泛应用,每年均有大量的PID致病新基因、新突变及新表型不断被发现和报道,IUIS-PID专家组亦形成每2~3年对PID分类进行更新的工作制度,相关文献分别在IUIS官网(http://www.iuisonline.org)和期刊上发表。
此外,美国Jeffrey Modell基金会根据PID的特点提出PID十大预警症状如下:(1) 一年内4次或以上新发耳部感染;(2)一年内2次或以上严重鼻窦感染;(3)使用抗生素2个月或更长时间收效甚微;(4)一年出现2次及以上肺炎;(5)婴儿体重不增或生长发育落后;(6)复发性深部皮肤脓肿或脏器脓肿;(7)持续鹅口疮或皮肤真菌感染;(8)需要静脉输注抗菌药物清除感染;(9)两次及以上深部感染,包括败血症;(10)有PID家族史。2018年Jeffrey Modell基金会定期统计全世界报道的各种类型的PID比例:单纯免疫球蛋白或抗体为主的缺陷占65%;联合免疫缺陷占6%;伴典型表现的联合免疫缺陷综合征占13%;免疫失调性疾病占3%,吞噬细胞和/或中性粒细胞缺陷占5%;而补体缺陷占5%,固有免疫缺陷及自身炎症性疾病占71%。因T辅助细胞功能低下,不能提供辅助B细胞合成分泌免疫球蛋白的信息,可能发生不同程度的抗体产生减少。因此在全部原发性免疫缺陷病中,超过80%存在不同程度免疫球蛋白和/或抗体缺陷。
病因和流行病学
根据2024 IUIS专家组数据表明,迄今已发现PID559种。PID发病率多通过病例登记的方式获得,这种方式可能漏算部分患者,因此存在报道的数据较实际数据低的可能。近年来,随着分子诊断的不断发展,新基因层出不穷,免疫缺陷病临床表型的大量拓展等原因,PID的发病率估计越来越高。目前认为,作为一个整体,PID或IEI的发病率介于1/(1000-5000)活产婴之间。我国目前无基于中国人群的PID患病率资料。根据整体的数据推断,如按照1/1000活产婴发病率推算,如2016-2018年,每年新生儿出生量为1600万至1800万,我国每年新增的PID患者16000-18000例;虽目前新生儿出生率下降,我国国家统计局公布2024年新生儿为954万,IEI新增量仍达到9540例。西方国家如美国最新公布的IEI的发病率为6/10000。实际上,在全世界,目前累积的IEI患者数量庞大。
由于PID是由单基因突变导致免疫细胞数量和/或功能异常的一类疾病,其发病机制基于不同病种,存在差异。生殖系基因突变、体细胞突变以及自身抗体产生等都可以导致免疫细胞数量或/和质量异常。生殖系基因突变指在生殖细胞中发生的任何可检测、可遗传的突变,它的遗传遵循孟德尔遗传规律,和体细胞突变最大的区别是能以不同遗传方式遗传给后代。例如,男性患儿IL2RG基因半合子突变导致X连锁重症联合免疫缺陷。体细胞突变指生殖细胞系列以外的细胞中发生的突变,也称作获得性突变。PID出现体细胞突变更常见的是嵌合突变,包括仅发生于体细胞嵌合体(仅仅表现为身体某些方面症状)、仅发生于生殖细胞的嵌合体和生殖细胞与体细胞嵌合体等类型。例如,先天性重症中性粒细胞减少症有生殖细胞嵌合体的报道,患儿父母无症状,外周血中性粒细胞水平正常,但基因分析发现父母有生殖细胞突变嵌合体。某些PID主要通过产生各种自身抗体发病。例如,抗I型干扰素抗体导致重症新冠感染,抗IL17自身抗体导致慢性皮肤黏膜念珠菌病等。
临床表现
由于病因不同,PID的临床表现极为复杂,但也有共性。因为这一类疾病的发病,是基于基因变异致免疫防御、免疫监视、免疫自稳三大免疫功能缺陷所致,因此,其共同的表现非常相似,即反复感染、自身免疫性疾病和易患肿瘤等。
1.反复和慢性感染 免疫缺陷最常见的表现是感染,感染有反复、严重、持久的特点。
临床上,患儿常需要持续使用抗菌药物,甚至使用抗生素预防感染的发生。
(1)感染的部位:PID可出现多部位感染,以呼吸道最常见,如复发性或慢性中耳炎、鼻窦炎、结膜炎、支气管炎或肺炎。其次为胃肠道,如慢性肠炎。皮肤感染可为脓疖、脓肿或肉芽肿。其他部位感染如脑膜炎和骨关节感染等,也可为全身性感染,如败血症、脓毒血症等。
(2)感染病原的特点:PID患者可出现不常见的和致病力低下的细菌感染。一般而言,抗体缺陷时易发生荚膜细菌感染。T细胞缺陷时易发生病毒(如疱疹病毒)、结核分枝杆菌和沙门菌属等细胞内病原体感染,也易发生霉菌和原虫感染。补体成分缺陷易发生奈瑟菌属感染。中性粒细胞功能缺陷时容易感染过氧化氢酶阳性细菌,如金黄色葡萄球菌。病原体的毒力可能并不强,呈机会性感染。
(3)感染过程的特点:PID感染常反复发作或迁延不愈,治疗效果欠佳,尤其是抑菌剂疗效更差,必须使用杀菌剂,剂量偏大,疗程较长才有一定疗效。部分患者必须预防使用抗生素才能减少感染反复发作。一些非免疫性因素也可能造成对感染的易感性,如呼吸道或泌尿道畸形、阻塞或发育异常、先天性功能异常等。在考虑PID时,应排除这些易患感染的非免疫因素。
2.自身免疫 PID可出现各种类型的自身免疫性疾病,最常见的为血液系统和内分泌系统受累等。具体包括溶血性贫血、免疫性血小板减少症、免疫性白细胞减少、系统性血管炎、系统性红斑狼疮、皮肌炎、免疫复合物性肾炎、1型糖尿病、免疫性甲状腺功能减退和关节炎等。
3.炎症性疾病 随着人们对PID认识的加深,还有一类疾病表现为炎症性疾病,包括反复或周期性发热、皮疹、骨骼肌肉症状如关节痛/关节炎、肌痛/肌炎为主要临床表现,此外还可累及消化、神经系统等,表现为腹泻、便血、惊厥、发育迟缓等,伴炎症指标明显升高,如C反应蛋白、血沉、血清淀粉样蛋白A等升高。
4.过敏 PID可出现各种类型的过敏反应,包括湿疹、皮疹、哮喘、食物过敏等。
5.淋巴增殖性疾病和肿瘤 PID可表现为淋巴增殖表现,表现为肝脾增大、淋巴结增大、脏器包块等。此外PID容易发生肿瘤,最常见的是白血病和淋巴瘤,其发生率在PID较正常人群高数十倍乃至100倍以上。淋巴瘤,尤以B细胞淋巴瘤(50%)最常见,T细胞瘤和霍奇金病、淋巴细胞性白血病、腺癌、鳞癌和其他肿瘤也可能发生。因此,临床上需重视病理活检,以鉴别是增殖期还是肿瘤期病变。
6.其他临床表现 某些PID除免疫功能异常致反复感染外,尚可有其他的临床特征,包括生长发育延迟或停滞、淋巴结肿大/缺如、特殊面容等。近来,PID伴发剧烈炎症性疾病和严重过敏性疾病的机制受到高度关注,比如家族性噬血淋巴组织细胞增生症,是因细胞毒缺陷导致炎症反应,诱发噬血现象;高IgE综合征可伴发严重过敏反应等。
辅助检查
免疫系统相互作用极为复杂,疑诊PID患者的辅助检查,重点是根据患者临床表现,初步判断患者可能属于哪一类PID,有针对性地检查。检查项目包括免疫学初筛、基因、基因突变体编码蛋白分析、基因突变体编码蛋白所在通路的功能分析、病原学分析、影像学分析等,必要时需完善骨髓细胞学、骨髓流式、PET-CT、组织活检病理等筛查肿瘤的可能。具体的辅助检查包括如下
1.免疫球蛋白测定 不同年龄正常儿童IgG、IgM、IgA和IgE值不同。免疫球蛋白水平在正常同龄儿均值±2S范围内视为正常。目前中国尚无不同年龄儿童Ig水平的正常值标准,因此根据不同年龄判断仍存在困难。年长儿和成人总Ig(包括IgG、IgM和IgA)大于6g/L者,应属正常,低于4g/L或IgG低于2g/L时提示缺陷。年长儿和成人总Ig为4~6g/L或IgG为2~4g/L者为可疑的抗体缺陷,应作进一步抗体反应试验或IgG亚类测定。IgG亚类包括:IgG1、IgG2、IgG3、IgG4,其在总IgG中的占比分别为70%、20%、7%和3%。不同年龄患者IgG亚类正常值不同,不同实验室的结果也不完全一致,最好应建立本地区和本实验室的正常参数值;此外,血清总的免疫球蛋白水平不一定能代表抗体反应能力,某些特殊疾病的血清免疫球蛋白水平正常,但抗原特异性抗体反应低下。抗体应答主要通过检测蛋白和多糖抗原诱导的IgG抗体应答。蛋白抗原IgG应答可采用:破伤风、白喉、流感嗜血杆菌B、肺炎蛋白连接疫苗等,亦可采用甲肝、乙肝疫苗。
2.抗A、抗B或抗AB同族凝集素 代表IgM类抗体功能,正常情况下,生后6个月婴儿抗A、抗B滴度至少为1∶8(AB血型者例外)。
3.外周血淋巴细胞亚群绝对计数 外周血淋巴细胞60%~80%为T细胞,因此外周血淋巴细胞绝对计数可代表T细胞数量,正常值为(2~6)×109/L;<1.5×109为可疑T细胞减少。一般而言,CD3+ CD4+细胞数<500/μl时可视为细胞免疫受损,<200/μl时则为严重缺陷。CD4/CD8比例<1时提示细胞免疫被抑制,当<0.3时,则为严重T细胞缺陷。B细胞在外周血淋巴细胞中占10%~20%,随年龄有一定变异。另外,淋巴细胞精细免疫分型检测可为免疫细胞亚型评估提供更为精确的参考。中国不同年龄及性别正常儿童外周血淋巴细胞各亚群比例及绝对计数详见参考文献9。
4.免疫器官筛查 婴幼儿期缺乏胸腺影者提示T细胞功能缺陷,但胸腺可深藏于纵隔中而无法看到,应仔细改变投射位置,以便暴露胸腺影。新生儿期常规胸部X线检查胸腺影,是筛查胸腺发育不全的重要手段。婴幼儿期发现淋巴结缺如,需警惕PID可能。
5.四唑氮蓝染料(NBT)试验和DHR实验 NBT为淡黄色可溶性染料,还原后变成蓝黑色甲颗粒。正常中性粒细胞进行吞噬时,糖代谢己糖磷酸旁路被激活,产生的氢离子和超氧根使NBT还原。预先用内毒素刺激中性粒细胞,或将NBT与乳胶颗粒混合后再进行中性粒细胞培养,涂片计数NBT阳性细胞数。正常人阳性细胞大于90%,而慢性肉芽肿病患者常低于1%。随着流式细胞术的应用普及,目前较多使用DHR呼吸爆发实验。DHR可自由透过活细胞膜进入细胞内,并被细胞内的ROS氧化,形成发绿色荧光的Rho123。临床采集50ul肝素钠抗凝外周血,去除红细胞,予佛波脂(PMA)刺激激活细胞后,DHR可形成发绿色荧光的Rho123,因此可通过流式细胞术检测中性粒细胞活化指标(活化百分比及平均荧光强度)。
6.补体CH50活性、C3和C4水平 总补体缺陷可被CH50活性法测定,其原理为血清补体成分能通过经典补体途径溶解抗体结合的羊红细胞,CH50正常值为50~100U/ml。C3占总补体的50%以上,C4是仅次于C3的主要补体成分。C3正常值新生儿期为570~1160mg/L,1~3个月530~1310mg/L,3个月~1岁620~1800mg/L,1~10岁770~1950mg/L。C4正常值为新生儿期70~230mg/L,1~3个月70~270mg/L,3~10岁70~400mg/L。
7.进一步检查 经过初步筛查,虽然一些PID已能做出诊断,但尚有一些疾病需进一步检查才能确诊。若基因分析发现突变体为已经报道致病突变,则可确诊。若突变体为新发突变,则需进一步通过功能验证明确突变体的致病性。突变体所在通路功能分析通常需要较高要求实验室进行。常见的功能实验如下:
T细胞增殖实验:外周T细胞在抗原、丝裂原、同种异体细胞和抗T细胞单克隆抗体的刺激下,发生增殖或克隆扩增是T细胞的重要功能之一。常用的T细胞刺激物为植物凝集素(PHA)、大刀豆素A(Con A)和美洲商陆(PWM)等。T细胞依赖的B细胞刺激物为PWM、多糖和抗原(PPD、细菌、病毒和霉菌),非T细胞依赖的B细胞刺激物为内毒素、抗免疫球蛋白、EBV、葡萄球菌蛋白A(SAC)和放线菌丝裂原。混合淋巴细胞培养(MLC,同种异体细胞DR)、抗原(PPD、细菌、病毒、霉菌)和超抗原(如葡萄球菌、肠毒素)刺激也是测定T细胞增殖的方法。T细胞增殖功能既往多采用3H-TdR掺入法,近年来多用荧光染料CSFE稀释法。该法以流式细胞术为基础,不仅避免使用同位素,还可采用特异性标记标识各种细胞亚群,观察其增殖能力。
B细胞活化和增殖功能:外周血单个核细胞在PWM和T细胞因子诱导下,B细胞表面MHC-DR表达能力、表面Ig类别的转换、B细胞增殖指数等实验可了解B细胞活化和增殖功能。加入各种调节因子于体外培养系统中,可了解影响其调控的各个环节。
补体成分及其活化片段测定:补体各成分及其调节蛋白的检测采用溶血或免疫反应法。经典途径激活时,C1、C4、C2、C3和C5明显下降;而旁路激活时C1、C4和C2正常,仅C3下降,但B、D和P因子则下降。
细胞毒性细胞功能:为了解淋巴细胞直接溶解靶细胞的能力,可测定细胞毒性功能,包括CTL、NK和ADCC功能。经典方法原理为将靶细胞用51Cr标记,在与患者淋巴细胞共同培养后,测定放射性释放量来代表靶细胞被溶解破坏的程度。细胞毒性囊泡中存在的CD107α分子在CTL和NK细胞与靶细胞接触时会短暂表达于细胞表面,因而在刺激NK细胞和CTL后,通过流式细胞术检测细胞表面CD107α水平变化可部分反映细胞毒功能。如明显上升说明细胞毒功能正常,如无上升则提示细胞毒功能缺陷,例如家族性噬血淋巴组织细胞增生症。
酶测定:腺苷脱氨酶(ADA)和嘌呤核苷磷酸酶(PNP)缺乏时,可测定红细胞内的ADA和PNP。测定羊水红细胞内该酶有助于产前诊断。
黏附分子测定:白细胞黏附分子缺陷(LAD)1型和2型见于白细胞增多症,反复软组织感染和趋化因子缺乏。LAD-1最常见,且伴有白细胞相关抗原-1(LFA-1)和整合素缺乏。LAD-2为白细胞选择素的配体缺陷,后者的功能为内皮细胞上的表皮生长因子与白细胞的黏附。采用流式细胞仪,可发现粒细胞缺乏CD11b(CR-3-Mac-1)和CD18(LAD-1),以及选择素配体缺乏(LAD-2)。
诊断
目前,PID诊断尚无统一标准,仅部分病种如重症联合免疫缺陷有诊断及管理指南。对有PID阳性家族史的患者,应立即启动免疫及基因筛查。临床表型、免疫学、实验室检查怀疑PID患者,应立即启动基因分析。基因明确是诊断PID的金标准。需注意的是,由于PID异质性很强,基因明确的PID应尽量个体化分析,常需实验室支撑,因此,很多地方无法实现,跨地区转诊对提高患者的诊治非常重要。
鉴别诊断
1.营养不良时的免疫缺陷、蛋白质营养不良和微营养素缺乏对免疫系统产生不良影响,包括细胞免疫、吞噬功能、补体等多方面受损。
2.药物引起的免疫缺陷,例如,B细胞抗体利妥昔单抗可使B细胞数量下降,也可影响补体依赖的细胞毒性作用等。联合使用其他免疫抑制剂,如环磷酰胺,可进一步增加感染风险,如巨细胞病毒感染、真菌感染等。
3.其他多种因素引起的继发性免疫缺陷,如严重感染后等,均有诱因去除后,免疫功能可恢复的特点。
治疗
1.一般处理 加强家庭宣教,早期诊断、早期识别、早期治疗,增强父母和患儿对抗疾病的信心等是提高PID患者生活质量及生存率最重要的因素之一。PID经正规治疗,是可以治愈的。
指导PID患者正确疫苗接种。严重抗体缺陷和细胞免疫缺陷患者,禁用活疫苗如脊髓灰髓炎口服疫苗、麻疹、腮腺炎、风疹和结核疫苗等,T细胞缺陷患儿不宜输血或新鲜血制品,以防发生移植物抗宿主反应。若必须输血或新鲜血制品时,应输注辐照血液制品。为防止巨细胞病毒(CMV)血源性感染,供血者应做CMV筛查。PID患儿最好不做扁桃体和淋巴结切除术,脾切除术视为禁忌。必须做脾切除者,应长期给予抗菌药物预防感染。糖皮质激素类应慎用。
2.替代治疗 即缺什么、补什么的治疗原则,可暂时性缓解临床症状。大约80%以上的PID伴有不同程度的低或无IgG血症。因此,伴有抗体缺陷PID是IgG替代治疗的绝对指针。其他替代疗法包括特异性免疫血清、输注白细胞、细胞因子等。
(1)静脉注射丙种球蛋白(IVIG):IgG替代治疗的基本方案为IVIG 0.4~0.6g/kg,每3~4周1次,维持5~6g/L的IgG谷浓度。治疗剂量应个体化,在遵循上述基本方案的基础上,目标剂量是保护该个体尽可能免受感染的剂量。此外需监测IVIG不良反应。IVIG的不良反应发生率低于2%,常出现于注射开始的头30分钟内,包括背痛、腹痛、头痛、寒战、发热和恶心。上述不良反应在减慢滴注速率后多能消失。有过敏史者,于注射前先给予抗过敏药物以预防不良反应的发生。极个别病例发生血压下降、呼吸困难等生命危象,应给予肾上腺素和糖皮质激素,并停止IVIG滴注。
(2)高效价免疫血清球蛋白:高效价免疫血清球蛋白是从免疫接种或自然感染的供体的血清中收集来的抗原特异性免疫血清,含有高效价特异性抗体。现正式用于临床的有水痘-带状疱疹、狂犬病、破伤风、乙肝以及巨细胞病毒的高效价免疫血清球蛋白。
(3)细胞因子治疗:IFN-γ治疗慢性肉芽肿病、IFN-γ缺陷和不全性IFN-γ受体缺陷病。粒细胞集落刺激因子(G-CSF)治疗先天性中性粒细胞减少症。胸腺素类包括胸腺五肽(TPS)等,对胸腺发育不全、湿疹血小板减少伴免疫缺陷病有一定疗效。
(4)酶替代治疗:腺苷脱氨酶(ADA)缺陷者,可接受ADA替代治疗。其可通过直接转换血浆内积累的腺苷脱氨酶底物,以及减少细胞内的有毒代谢产物,从而纠正ADA缺陷所致的代谢紊乱。迄今为止,全球已有超过150人接受替代治疗。患者可很好耐受,可修复免疫系统至防护水平,但长期随访提示免疫系统的恢复仍不完全。
3.抗感染及预防感染治疗 PID患者容易发生感染,常需静脉使用抗生素。严重患者需早期、联合静脉使用抗生素。应基于患者免疫学特点分析易感病原菌,从而经验性地选择抗生素治疗。应尽早寻找病原学依据,完成药敏试验,尽量根据药敏试验结果调整抗生素。若治疗过程中病情难以控制,还需警惕细菌耐药的可能。耶氏肺孢子菌肺炎(PCP)是细胞免疫缺陷病和HIV感染的重要并发症,当CD4+细胞计数1岁内婴儿<1500/ml,1~2岁<750/ml,2~5岁<500/ml,年长儿<200/ml,或任何年龄组CD4细胞<25%总淋巴细胞时应进行感染的预防。此外,T细胞缺陷和粒细胞缺陷的部分患者,还需预防抗真菌治疗。
4.PID的免疫重建(immune reconstitution) 免疫重建是采用正常细胞或基因片段植入患者体内,使之发挥其功能,以持久纠正缺陷。免疫重建的方法有胸腺组织移植、干细胞移植和基因治疗。
(1)造血干细胞移植(hematopoietic stem cell transplantation, HSCT):包括骨髓、外周血和脐血来源的造血干细胞移植。自1968年首次采用HSCT成功治疗重症联合免疫缺陷病(severe combined immunodeficiency, SCID)来,全球已有数千例PID患儿接受了骨髓移植,并成为多种PID的重要根治手段。根据供者情况不同,HSCT分为同种异体同型合子HSCT、同种异体半合子HSCT(常为家庭成员父母或兄弟)和无关供体HSCT等。近年来,预处理方案的改进、支持治疗、移植并发症处理技术等使HSCT治疗PID的远期疗效得到明显提高。
(2)基因治疗:是将正常的目的基因片段整合到患者干细胞基因组内,使其能在患者体内复制而让正常基因能持续在体内表达。理论上讲,凡是能通过造血干细胞移植治疗的PID都有基因治疗的指针。逆转录病毒和慢病毒是截至目前最常用的病毒转染载体,分离造血干细胞,体外转染带有正常目的基因的载体病毒,然后输入患者体内。基因治疗PID自1990年治疗第一例腺苷脱氨酶缺乏症严重联合免疫缺陷患儿以来,已取得一定成效,但既往由于病毒载体本身可能导致T细胞白血病,应用受到限制。近年来,随着新一代自灭活载体的不断发展,基因治疗安全性得到大幅度提高。目前使用基因治疗的PID包括重症联合免疫缺陷、慢性肉芽肿病等多种疾病。
诊疗流程(图66-1)
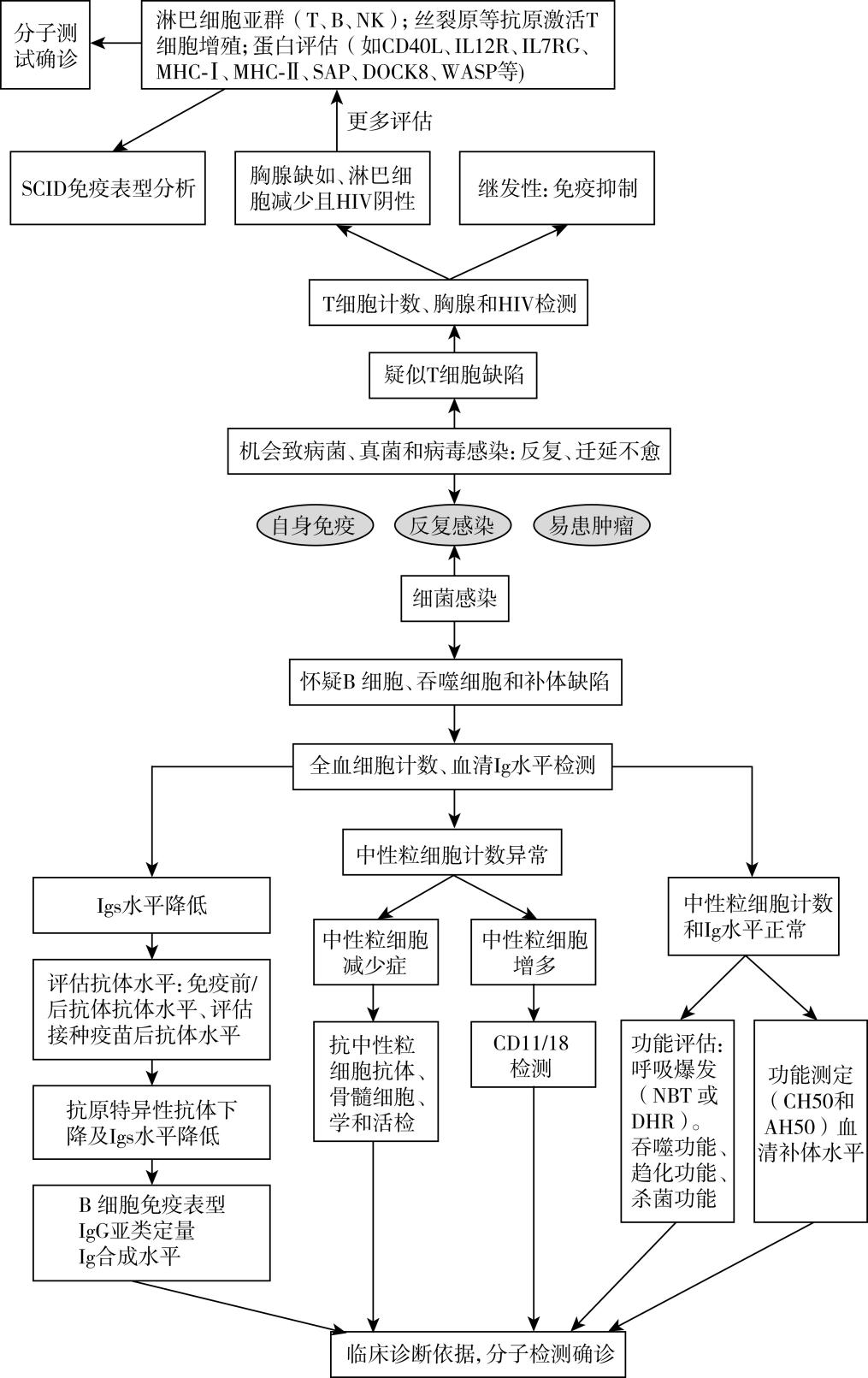
图66-1 原发性免疫缺陷病诊疗流程
参考文献
[1] M. Cecilia Poli, Ivona Aksentijevich, Ahmed Aziz Bousfiha, et al. Human inborn errors of immunity: 2024 update on the classification from the International Union of Immunological Societies Expert Committee. Journal of Human Immunity. Journal of Human Immunit,2025, 1(1): e2025003.
[2] Akalu YT, Bogunovic D. Inborn errors of immunity: an expanding universe of disease and genetic architecture. Nat Rev Genet,2024,25(3):184-195.
[3] ischer A. Gene therapy for inborn errors of immunity: past, present and future. Nat Rev Immunol,2023,23(6):397-408.
[4] Deng M, Mao H. Inborn errors of immunity inmainland China: the past, present and future. BMJ Paediatr Open,2023,7(1):e002002.
[5]赵晓东.儿童免疫学.2版.北京:人民卫生出版社,2022.
[6]毛华伟,孙金峤,李冀,等,原发性免疫缺陷病免疫球蛋白G替代治疗专家共识,中华儿科杂志,2019,57(12):909-912.
[7] Marzieh Tavakol1, Mahnaz Jamee, Gholamreza Aziz. Diagnostic approach to the patients with suspected primary immunodeficiency, endocrine, metabolic & immune disorders - Drug Targets, 2020, 20, 157-171.
[8] Vicki Modell1,Jordan S. Orange1,Jessica Quinn1, Fred Modell1, Global report on primary immunodeficiencies: 2018 update from the Jeffrey Modell Centers Network on disease classification, regional trends, treatment modalities, and physician reported outcomes, Immunol Res,2018,66(3):367-380.
[9] Yuan Ding , Lina Zhou, Yu Xia, et al. Reference values for peripheral blood lymphocyte subsets of healthy children in China, J Allergy Clin Immunol. 2018 ,142(3):970-973.