


162、早老症
罕见病诊疗指南(2025年版)
概述 早老症(Hutchinson-Gilford progeria syndrome,HGPS),也称儿童早衰症,全称哈钦森-吉尔福德早年衰老综合征(Hutchinson-Gilford progeria syndrome,HGPS),最早在1886年和1897年分别被Jonathan Hutchinson医生和Hastings Gilford医生所报道。HGPS是一种极为罕见的严重的常染色体显性遗传病,由核纤层蛋白A基因(LMNA)第11号外显子或者第11号内含子突变引起,这些突变位点可产生早老蛋白(progerin),早老蛋白在体内积累最终导致疾病。临床上以加速性衰老为其显著的特征,以皮肤硬肿和生长迟缓为其首要就诊原因,病变可累及多系统,但智力正常,HGPS可迅速发展为心功能不全、血管硬化、心脏瓣膜钙化和冠状动脉疾病,患者大多死于心血管疾病,HGPS患者的平均寿命仅14.6岁。
病因和流行病学
2003年美国科学家Collins及其团队和法国科学家同时发现核纤层蛋白A基因(LMNA)是HGPS的致病基因。LMNA基因位于染色体1q21.2,包含12个外显子和11个内含子,通过第10号外显子中的可变剪接编码四种A型核纤层蛋白(A、C、CΔ10和C2),其中核纤层蛋白A(lamin A)和C(lamin C)是最普遍表达的,但不存在于未分化细胞中,是细胞骨架的重要组成部分,由于其特殊的结构和定位,核纤层蛋白参与了多种细胞生物过程:调节细胞衰老及分化、DNA损伤反应、维持基因组稳定性、参与细胞转录调控、信号传导、异染色质重塑等.
正常情况下LMNA基因12个外显子均被转录产生初级RNA转录物 Prelamin A,PrelaminA由664个氨基酸组成,其C末端有一个“CAAX盒”(C半胱氨酸、A脂肪族氨基酸、X任意一种氨基酸)。首先由“CAAX盒”中的半胱氨酸开始被蛋白法尼酰基法尼基化;之后剩余的C末端“AAX”被锌金属蛋白酶ZMPSTE24(zinc metalloproteinase Ste 24)切除;当AAX被切除后,之前剩余的法尼半胱氨酸被内质网中的异戊烯基半胱氨酸羧基甲基转移酶(isoprenylcysteine carboxyl methyltransferase,ICMT)甲基化;最后由ZMPSTE24切除PrelaminA末端最后的15个氨基酸,切除之后释放出成熟的含646个氨基酸的LaminA。
根据致病基因突变位点的不同,HGPS可分为经典型HGPS和非经典型HGPS。经典型HGPS是LMNA基因经典位点c.1824C>T(p.Gly608Gly)杂合突变所导致,该点突变后可以激活11号外显子中一个隐秘的剪接位点,导致C末端附近50个氨基酸的缺失,阻止Zmpste24的第2次切割,进而导致缩短的、永久法尼基化异常的prelamin A的积累,这种异常的prelamin A称为早老蛋白(progerin),该蛋白结合至核纤层中并产生多种毒性,导致在细胞水平上细胞核膜结构及功能产生显著缺陷。非经典型HGPS是LMNA基因第11外显子或第11内含子区域具有产生早老蛋白的突变位点。另外,除第11外显子/第11内含子区域的突变外,LMNA基因其他位置的突变也可能导致过早衰老的表现,这类疾病称为早衰样核纤层蛋白病(progeroid laminopathies,PL),这些突变不产生早老蛋白,但同样会导致与HGPS类似的疾病表现,如LMNA R527C是PL的热点突变位点,R527位于蛋白质结构域的外表面并参与形成盐桥,此位点突变后会破坏蛋白质的表面结构,对核纤层结构产生影响,最终导致PL发病,在此类PL患者细胞中,并未发现有prelaminA和早老蛋白的积累,另外,LMNA基因的T528M和M540T复合杂合突变也并没有早老蛋白的堆积。
此外,锌金属蛋白酶24(zinc metalloproteinase 24,ZMPSTE24)突变也可导致PL,其临床表现与经典型HGPS非常相似,ZMPSTE24基因突变也导致异常的prelamin A,但不是早老蛋白。
HGPS呈散发分布,极为罕见,根据美国早衰症研究基金会(Progeria Tesearch Foundation, PRF)统计,HGPS的患病率约1/2000万,没有性别或种族差异;全球每400万~800万例新生儿中仅1例患有该病,估计全球病例数为 400~450 例,截至2023年3月31日我国登记在册的HGPS患者人数20人,PL患者人数26人。
临床表现
HGPS儿童通常在出生时表现正常,生后不久,特别是生后1年内逐渐开始呈现衰老征象,具体如下:
1.皮肤和毛发 皮肤和毛发异常通常是HGPS的初始症状,一般在12个月前就明显。最常见的特征为皮肤硬皮样改变、突显的浅表脉管系统、色素沉着和脱发。有研究报告约78%的HGPS患儿有皮肤硬皮样改变,常常累及腹部和双侧下肢,初次出现的平均年龄为1.9个月,24%患儿在出生时就出现了这些变化。突出的浅表脉管系统则表现为口周紫绀和头皮或/和四肢、躯干有显露静脉。在硬皮样变化区域观察到色素减退和色素沉着过度。
所有HGPS患儿虽然出生时有正常的毛发,但头几年内头发逐渐脱落(一般从颞部和枕部开始),只留下稀疏的“绒毛样”头发。眉毛也会在生后最初几年发生脱落,留下非常稀疏的浅色眉毛,而睫毛通常稀疏。所有正常的头发脱落后,指/趾甲通常生长缓慢,易长成异常形状,有时会发生裂开,但不会影响手足功能。
2.生长迟缓 生长迟缓是HGPS常见的首诊原因。HGPS患儿在胎儿期和生后早期发育一般都正常。出生时没有明显症状,可能与胎儿早期发育过程中细胞未完全分化时缺乏早老蛋白有关。生后第一年,尤其是生后三月后出现明显生长迟缓,表现为体重不增甚至下降、身高不长以及全身性脂肪萎缩。有研究显示HGPS患儿在2月龄时体重即下降到同年龄同性别健康儿童生长曲线第3百分位以下,15月龄时身高低于同年龄同性别健康儿童生长曲线第3百分位。HGPS患儿头围也常常低于正常同龄儿童第3百分位。
3.颅面部和口腔 HGPS患儿典型的颅面特征包括脱发或头发稀疏、颅面不对称、头骨变薄、眼睛突出、鼻梁狭窄、中下面部比例过小、面部脂肪缺乏和头皮静脉显露。60%~70%的患儿呈现尖状腭弓。约50%患者舌系带短而厚,从而导致舌部活动受限。狭窄的气道和僵硬的喉部结构导致高音。几乎所有的HGPS患儿都有口腔及牙齿异常,这可能与患儿下颌骨发育明显延迟及牙齿发育异常有关。下颌骨发育延迟引起小下颌,进一步影响牙萌出延迟、牙齿拥挤和咬合不良;颞下颌关节活动受限而导致HGPS患儿开口困难;与此同时,HGPS常表现为轻到中度牙周炎,最常缺失的恒牙是第二前磨牙和侧门牙。放射学检查结果表明牙冠和牙根发育不规则。
4.骨骼和关节 所有HGPS患儿都有不同程度的骨骼异常和关节畸形,如进行性髋外翻,这类HGPS特有的骨骼发育不良跟营养不良无关,也不同于老年人骨质疏松。髋关节发育不良通常是进行性的,可能导致缺血性坏死、髋关节移位和无法承重。骨骼受累可导致特征性面部外观伴小下颌或下颌内缩,牙列拥挤,鼻梁狭窄,身材矮小。X线下可表现为髋外翻、髋关节发育不良、缺血性坏死、肢端骨溶解、远端指骨溶解、锁骨小、锁骨远端吸收、肋骨薄、长骨远端干骺端矿化减少等。梨状胸结构和小锁骨可导致可复位的肩关节脱位。
韧带和皮肤变化引起的关节挛缩可限制运动范围,包括手指、肘部、臀部、膝盖和脚踝在内的多个关节挛缩可能在出生时和/或以后出现。与同龄人相比,HGPS患儿的骨骼较小,骨密度通常处于中等或偏低的水平。但自发性骨折或骨折的频率并没有明显增高,当发生骨折时,骨骼也愈合良好。
5.心血管系统 心力衰竭是HGPS患儿死亡的首要原因。尽管HGPS患儿血脂紊乱并不明显,但是他们仍会发生严重的动脉粥样硬化。左室舒张功能障碍是HGPS患儿所有年龄组中最常见的超声心动图异常,而且该患病率随年龄增长而增加。其他心脏异常,包括心肌梗死和主动脉瓣狭窄或反流以及左心室肥厚,一般在10多岁后发生。收缩期功能障碍通常出现在疾病后期。心绞痛、劳累时呼吸困难或明显心力衰竭出现在病程末期。
6.脑血管系统 卒中或其他脑血管疾病占HGPS儿童死因的10%左右。任何年龄都有发生脑血管事件的可能,脑血管病变通常表现为卒中或短暂性脑缺血发作,以5~10岁常见,最早可发生在4岁,可表现为一侧或双侧面部、手臂或腿突然麻木或无力,构音不全,常伴有头痛、癫痫发作、肢体无力,少数患儿以精神情绪异常为主要表现。
7.听力和眼睛 传导性听力损失在HGPS人群所有年龄段都非常普遍,低频听力损失比高频听力损失更普遍。夜间睁眼症(睡眠时无法完全闭上眼睛)很常见。因此,可能出现角膜干燥和混浊。在少数个体中,角膜溃疡是由于暴露性角膜炎引起的。
8.其他方面
(1)约50% HGPS个体会发生胰岛素抵抗,甚至发生2型糖尿病。
(2)约37.5% HGPS个体有脂肪肝表现;约75% HGPS个体血高密度脂蛋白(HDL)较正常值低。
(3)智力发育正常。
(4)免疫功能正常。
(5)肾功能、神经认知功能正常。
(6)在HGPS患儿中肿瘤发生率并不比正常人群中的肿瘤发生率高。
(7)其他正常衰老的特征如老年人格变化、Alzheimer病、老花眼、白内障等在HGPS患儿中都没有报道。
(8)本病患儿血小板通常升高,凝血功能大致正常。
(9)性腺不发育或性发育明显迟缓。大约40%的患者停留在Tanner 1期,其余患者可发展到2期,其特征是长出稀疏的阴毛和/或乳芽。据估计60%的HGPS女性患者会经历月经初潮,月经初潮的平均年龄与健康女性月经初潮平均年龄并无显著差异。
辅助检查
1.营养评估和膳食调查 绘制体重和身高曲线评估患儿生长发育情况,调查患儿膳食摄入情况,指导及调整患儿饮食。应用双能X线检查评估人体成分组成(脂肪含量、脂肪分布、肌肉含量等)和骨密度;HGPS患儿的脂肪分布显著异常、肌肉含量也较正常同龄儿低。在评估骨密度时需按照身高-年龄来校正Z评分。
2.内分泌代谢评估 定期监测HGPS患儿血葡萄糖、血清胰岛素、血脂、甲状腺功能及肝脏超声检查等。胰岛素抵抗指数通过胰岛素抵抗稳态模型评估(HOMA-IR):空腹胰岛素(mU/L) ×空腹血糖(mmol/L)/22.5,并定义HOMA-IR>3为胰岛素抵抗(IR)。
3.心电图和超声心动图 HGPS患儿心脏受累心电图可表现为长QT间隔、双心室肥大和双心房肥大、较深的Q波,ST-T波和较短的PR间隔等。超声心动图用于评估心室和瓣膜功能,HGPS患者最普遍的超声心动图异常是左心室舒张功能障碍,其他超声心动图表现为左心室肥厚、左心室收缩功能障碍和瓣膜疾病(主动脉瓣和主动脉瓣膜增厚、主动脉瓣和二尖瓣狭窄和反流等)。
4.颈动脉超声检查 颈动脉超声检测颈动脉内膜厚度,是否存在狭窄阻塞,是否存在动脉粥样斑块等。
5.头颅MRI/MRA 定期行头颅MRI/MRA及颈部MRA检查评估HGPS患儿头颅及头颈部血管情况。HGPS儿童常发生脑部慢性灌注不足,易引起的脑白质缺血性损伤。HGPS儿童脑梗死主要分布在大血管领域,涉及所有的血管区域,并倾向于颈内动脉、大脑中动脉和分水岭区域,大脑后动脉狭窄少见。
6.骨骼及关节X线检查 可有以下异常表现:
(1)肢端骨质溶解:远端指/趾骨的骨吸收是HGPS最早的影像学表现。
(2)下颌骨发育不良:小颌畸形和下颌后缩。
(3)锁骨吸收:锁骨远端有骨溶解。
(4)肋骨变细变尖:肋骨细,末端逐渐变尖。
(5)钟形胸:肋骨呈“下垂”状,胸尖锥形,使胸部呈“钟形”或“金字塔形”。
(6)髋外翻畸形:股骨颈轴度异常增大(> 125°)。
(7)短髋畸形:股骨颈短而宽。
(8)髋膨大畸形:股骨头大而宽,呈非球面状。
(9)髋臼发育不良:髋臼异常浅,导致承重受限,髋关节半脱位,运动范围丧失和骨关节炎。
(10)股骨头缺血性坏死:当股骨头失去适量的血液供应,会导致扁平、破碎和软骨下塌陷。
(11)长骨畸形:长骨细长,干骺端宽大呈放射状(肱骨近端,股骨远端,胫骨近端),骨骺大而宽,骨干矿化正常,干骺端和骨骺脱矿质。
(12)肱骨远端肱骨小头增大:肱骨远端外侧的生发中心明显增大。
(13)心血管和软组织钙化:心血管及腹部软组织或四肢指端毛细血管周围簇可能出现钙化。
7.眼部检查 HGPS患儿眼部检查可发现角膜变薄、角膜破裂、持续角膜溃疡、侵袭性翼状胬肉和继发性角膜瘢痕等,一般无斜视、白内障、青光眼或视网膜色素变化。Oculus眼表综合分析可发现睑板腺分泌功能差,眼底检查可发现眼底动脉/静脉迂曲。
8.听力检查 建议对所有HGPS患儿进行声导抗及耳声发射测试,对5周岁以上的HGPS患儿给予纯音听阈测定。大部分HGPS患儿存在不同程度的以低频损失为主的传导性听力损失。对中重度听力损失的HGPS患儿行耳内镜检查,可发现外耳道狭窄、鼓膜内陷、镫骨头消失、光锥消失等特征;行颞骨CT检查则可发现乳突气房气化欠佳,颞骨骨质疏松等。
9.智力评估与社会适应能力评估 使用韦氏儿童智能量表检测一般智力水平,使用婴儿-初中生社会生活能力量表评估0.5~14岁儿童适应性能力,HGPS患儿智力与社会适应能力评估一般正常范围。
10.基因检测
(1)单基因检测靶向性分析:拟诊HGPS患者是否存在LMNA c.1824C>T突变位点,约90% HGPS患者为该经典突变类型;如果在靶向分析中未发现致病性突变,则可进行LMNA序列分析,应包含LMNA全部外显子和内含子。
(2)多基因Panel检测:多基因panel应包括LMNA、ZMPSTE24和其他可能的致病基因。该种检查是识别疾病遗传突变基因性价比比较高的方法,但会限制识别不确定意义的变异和基因中的致病性变异。
(3)全面的基因组测序:当患儿的临床表型与其他早衰疾病无法鉴别区分时,则建议选择全面的基因组检测。一般首选全外显子测序,也可选择全基因组测序。
诊断
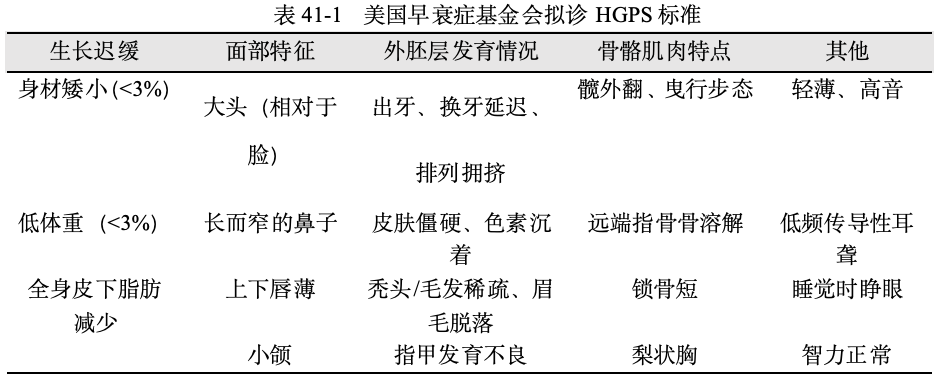
1.临床诊断 如果患儿1岁之后出现严重生长迟缓、皮肤硬皮样改变、脱发、全身性脂肪营养不良、后颌畸形、锁骨远端吸收、指骨末端吸收、髋外翻、乳牙延迟/不完全萌出,但智力发展正常,则可以临床诊断为HGPS。美国早衰症基金会提出了HGPS拟诊标准,详见表41-1。
2.确诊标准 通过典型的临床表现结合基因检测即可明确做出诊断。
经典型HGPS的致病基因:LMNA基因第11外显子c.1824C>T(p.Gly608Gly)杂合突变。
非经典HGPS的致病基因:LMNA基因第11外显子区或第11内含子中具有产生早老蛋白的杂合突变位点(但除外第11外显子c.1824C>T杂合突变)。
鉴别诊断
常见的需要与HGPS鉴别诊断的疾病包括:
1.成人早衰症(Werner syndrome,WS) WS是一种染色体隐性方式遗传的早衰的疾病,由WRN基因突变引起。WS个体正常发育至青春期,然后在青春期生长停止,患者成年后通常比平均身高矮。通常在20多岁时观察到头发脱落/变白、声音嘶哑和硬皮病样皮肤改变,随后在30多岁时出现双侧眼白内障、2型糖尿病、性腺功能减退、皮肤溃疡和骨质疏松症。心肌梗死和癌症是最常见的死亡原因。
2.不典型成人早衰症(atypical werner syndrome,AWS) AWS是一种常染色体显性遗传病,部分由LMNA突变引起,主要发生LMNA第5号(20%)或第2号外显子(50%)。AWS患者通常在儿童时期发育正常,但通常由于青少年时期缺乏青春期生长突增而停止生长,因此缺乏生长突增和身材矮小是AWS最常见的首要症状,约60%的AWS患者在10~20岁之间开始出现初始临床症状,可能比CWS患者更早出现。
3.下颌骨-骶骨发育不良(mandibuloacral dysplasia,MAD) MAD是一种罕见的常染色体隐性遗传疾病,以早衰合并脂肪营养不良、骨骼发育异常为其主要的临床特征。根据基因突变的不同分为两大类:第一类是MADA,由LMNA基因突变引起,合并A型脂肪营养不良;第二类是MADB,由ZMPSTE24基因突变引起,合并B型全身性脂肪营养不良。MADA和MADB的临床症状通常出现在儿童早期(4~5岁),伴有骨骼、皮肤和脂肪组织改变,其中MADB的体征和症状可在2岁时出现,临床表型也比MADA更严重。MAD的临床表型为(1)颅面异常(下颌发育不全、牙齿过度拥挤、喙状鼻子和突出的眼睛);(2)骨骼畸形(肢端骨质溶解、关节僵硬、锁骨发育不全);(3)皮肤变化(色素沉着过度、硬皮病样、脂肪营养不良);(4)早衰样综合征(生长发育迟缓、身材矮小、特殊面容、高音调、脱发、皮肤萎缩和指甲发育不良),虽然MAD与HGPS许多临床症状一致,但两者有所不同,HGPS患者常见的死亡原因是心脏衰竭、动脉粥样硬化等,而MAD患者很少发现心脏功能不全。
4.限制性皮肤病(restrictive dermopathy,RD) RD是一种罕见的、致命的常染色体隐性遗传核纤层蛋白病。可以是由LMNA基因或ZMPSTE24基因突变引起,但大多数因纯合或复合杂合ZMPSTE24突变所致。RD患儿产前即有异常表现如羊水过多和胎动减少,且早产出生几乎是普遍的。出生时体型小是另一个特点。特征性的临床表现有紧绷、脆弱的皮肤、浅表糜烂或溃疡以及明显的浅表脉管系统。典型的面部特征包括“挤压”的鼻子、低垂的耳朵、小颌或小“O”样嘴。患儿还常常合并多发关节挛缩。一系列临床症状常常导致新生儿早产并在出生后几天或几个月内死亡,至今为止,RD是最严重的核纤层蛋白疾病。
5.非典型早衰综合征(atypical progeroid syndromes,APS) APS与上述四种类型的早衰样核纤层蛋白病存在很多相似点,也由LMNA基因或ZMPSTE24基因突变引起,这些突变可以是显性或隐性的,并改变整个蛋白质结构中的残基,引起细胞核层损伤,该损伤与产生早老蛋白的等位基因产生的损伤重叠,临床症状可以很轻也可以很严重,其特征均为生长迟缓,受累器官与经典HGPS相同(骨骼、体脂肪、皮肤和头发),但发病年龄不同。
6.Cockayne综合征(Cockayne syndrome,CS) CS是由于ERCC6或ERCC8的双等位基因致病变异导致,可呈现多种快速衰老的临床特征,包括恶病质侏儒症、严重的神经系统表现、小头畸形和认知缺陷、色素性视网膜病变、白内障、感音神经性聋以及行走和喂养困难等,平均寿命约12岁。
7.Néstor-Guillermo早衰综合征(Néstor-Guillermo progeria syndrome,NGPS) NGPS是一种罕见的常染色体隐性遗传病,由BANF1基因纯合突变所致,常在2岁以后发病,NGPS的临床表现与HGPS相似,包括面容老化、生长迟缓、皮下脂肪减少、四肢细弱和关节僵硬等。其余特征包括脂肪萎缩、骨质疏松症、严重的骨质溶解。此类型的早衰症患者没有心血管疾病以及代谢综合征的相关症状,平均寿命也长于其他早老症,并且基因检测并没有LMNA或ZMSPTE24基因的突变或缺失。
治疗
针对HGPS的治疗可分为对症治疗、特异性治疗和基因治疗。
1.对症治疗
(1)生长发育迟缓:少量多餐,进食高热卡零食以尽量增加热卡的摄入。
(2)牙齿问题:严重的牙列拥挤和阻碍牙齿萌出可能需要拔牙治疗;加强口腔护理,早期使用含氟牙膏、漱口水以预防龋齿的发生。
(3)皮肤问题:户外活动时,建议在包括头部在内的所有皮肤暴露部位使用防晒霜。
(4)骨骼及关节异常:一般情况下应鼓励患儿进行适当负重活动(例如步行,奔跑,跳跃),这有益于维持骨密度水平。同时为了保持最佳的骨骼健康,患儿在饮食中需接受充足的钙和维生素D的摄入,但不建议补充钙剂。一旦患儿出现骨关节炎,则其初始治疗包括以恢复运动和肌肉力量的物理疗法和减轻疼痛的消炎药物治疗为主;晚期髋骨关节炎患儿需要助行器辅助,当患儿无法独立行走时需要坐轮椅;随着关节炎进展,可以考虑手术替代方法重建受累关节,以形成稳定协调的关节。
(5)心脑血管疾病:规律健康饮食及适当运动是前提,同时注意避免贫血、高热及脱水。同时建议使用低剂量阿司匹林以预防卒中和心脏病发作。一旦患儿出现心脑血管功能下降的迹象或症状,如高血压、短暂性脑缺血发作、卒中、癫痫、心绞痛、运动时呼吸困难、心电图变化、超声心动图变化或心脏病发作,需要根据情况给予降血压、强心、加强抗凝等治疗。
(6)眼部问题:暴露性角膜炎可在白天用眼部润滑液治疗,在睡眠时用保湿软膏治疗或用皮肤胶带封闭眼睑。
(7)听力问题:低频传导性听力损失往往不干扰日常生活活动,如果临床需要则可以使用助听器。
2.特异性治疗 HGPS特异性治疗策略主要包括:①降低异丙烯化和甲基化早老蛋白的毒性水平;②抑制prelamin A mRNA的异常剪切;③诱导清除早老蛋白;④减少与早老蛋白积累相关的有害下游效应,上述的治疗策略目前绝大多数仅限细胞、动物实验层面。
洛那法尼(lonafarnib)是当前唯一可及的治疗早老症及其他的加工缺陷型早衰样核纤层蛋白病的特异性药物,它是法尼基化转移酶抑制剂,可降低异丙烯化和甲基化早老蛋白的毒性水平,目前我国可通过管制下用药计划(MAP)申请用药。Zoledronate/pravastatin可协同作用于法尼基焦磷酸的合成途径,Ⅱ期单中心单臂临床试验结果显示,zoledronate/pravastatin可改善HGPS患儿的体重和骨密度,没有严重副作用;但是在另一项临床试验lonafarnib联合zoledronate/pravastatin治疗HGPS的结果显示,与lonafarnib单药治疗相比,HGPS患儿骨密度增加,但其他方面并没有改善。
everolimus:是一种mTOR抑制剂(西罗莫司类药物),可增加细胞自噬,进而诱导清除早老蛋白。该药已被批准为治疗晚期肾母细胞癌等肿瘤性疾病。西罗莫司通过增加自噬作用改善HGPS患者成纤维细胞的细胞表型,并延长lamin A缺陷小鼠模型的寿命。目前美国正在进行lonafarnib联合everolimus治疗HGPS的临床试验。
remodelin:是一种小分子药物,通过抑制N-乙酰转移酶10(NAT10)改善了HGPS的细胞缺陷,有效治疗了HGPS小鼠的心脏疾病,包括主动脉外膜纤维化的减少、挽救了血管平滑肌细胞和主动脉及冠状动脉中的平滑肌肌动蛋白的损失,显著增强了早老小鼠的寿命。remodelin将进一步进行与lonafarnib的联合疗法的临床试验。
3.基因治疗 基因治疗是唯一可能根治HGPS的方法。研究者曾尝试采用CRSIPR-Cas9技术敲除LMNA基因的致病拷贝,但这有破坏LMNA基因野生型拷贝的风险,难以运用于临床。2017年David Liu团队研制了可实现A.T—G.C碱基对转换的新型单碱基编辑器ABE;他们随后的研究证实了ABE在HGPS治疗中的巨大潜能:在低脱靶风险的条件下,ABE能直接逆转早衰小鼠模型中的致病点突变并明显改善模型的多种症状,且单次注射ABE病毒便能显著延长其寿命。当然这些基因治疗还存在很大的局限性,包括在染色体水平上的脱靶效应,以及与病毒传递系统相关的副作用(基因整合,免疫系统诱导等),临床应用还任重道远。
4.干细胞治疗 间充质干细胞具有多向分化和无限增殖等功能,同时还具有调节免疫反应、易获取、来源广泛等特点。近年来应用于各类疾病的治疗中。2016年我国报道了首例运用胎盘间充质干细胞治疗HGPS的临床研究。14 岁早衰症女孩接受了胎盘间充质干细胞治疗,病情得到了缓解,听力和肝功能得到了改善。
并发症监测
由于HGPS患儿往往合并多种问题,因此定期检查非常重要,主要包括以下几方面:每半年进行心电图、心脏超声以及颈动脉超声检查;每年进行神经系统评估、颈部和头部MRI/MRA、血脂分析、口腔和牙的检查、听力及眼睛的检查、骨骼和关节的评估、运动能力评估等。
诊疗流程(图41-1)
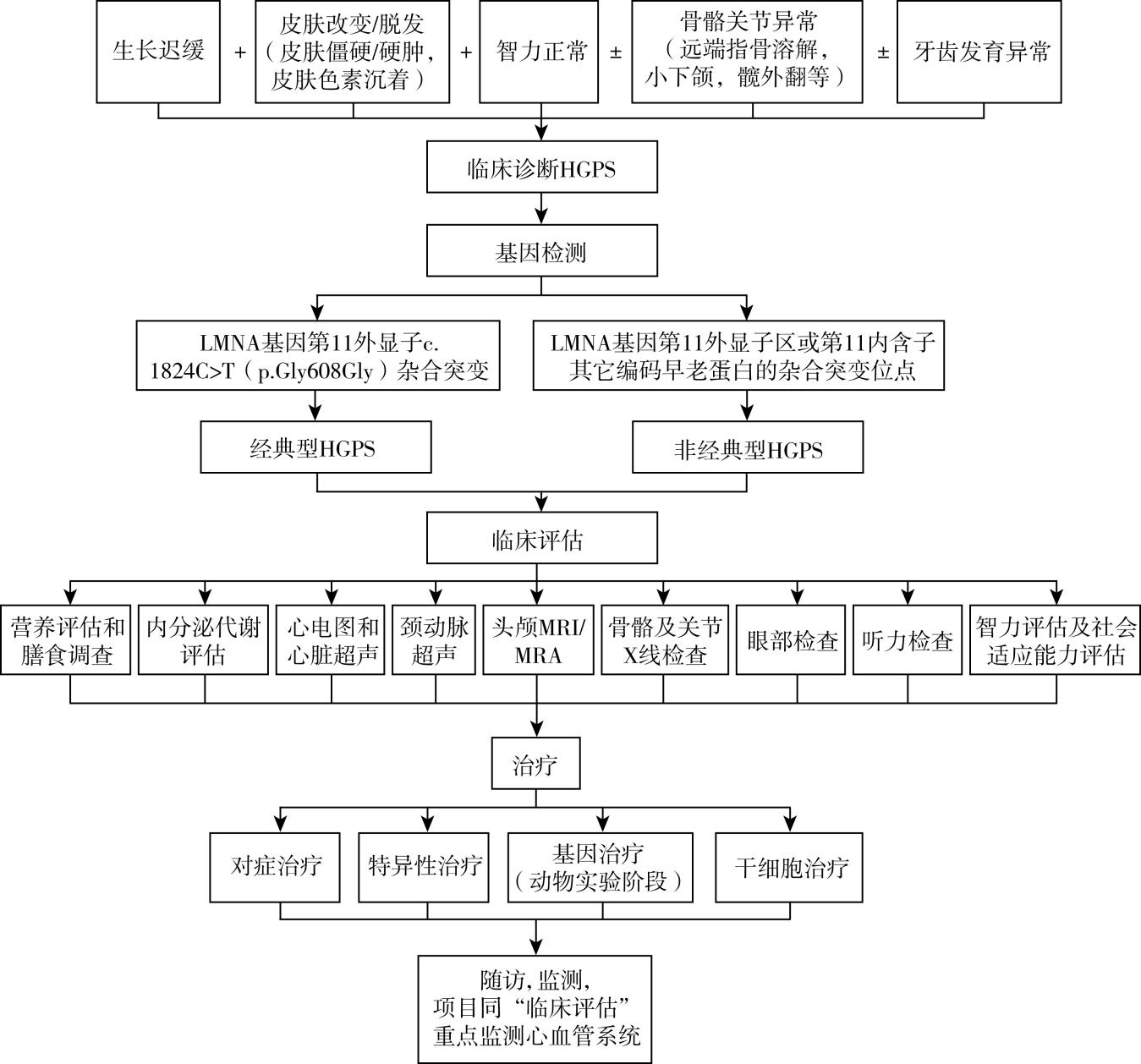
图41-1 早老症的诊治流程
参考文献
[1]Cisneros B, García-Aguirre I, De Ita M, et al. Hutchinson-Gilford Progeria syndrome: cellular mechanisms and therapeutic perspectives. Arch Med Res,2023,54(5):102837.
[2]Janota CS, Calero-Cuenca FJ, Gomes ER. The role of the cell nucleus in mechanotransduction. Curr Opin Cell Biol,2020,63:204-211.
[3]Gordon LB, Brown WT, Collins FS. Hutchinson-Gilford Progeria syndrome//ADAM MP, FELDMAN J, MIRZAA GM, et al. GeneReviews(®). Seattle (WA): University of Washington, Seattle Copyright © 1993-2024, University of Washington, Seattle, 1993.
[4]Marcelot A, Worman HJ, Zinn-Justin S. Protein structural and mechanistic basis of progeroid laminopathies. Febs j,2021,288(9):2757-2772.
[5]李东明. LMNA R527C隐性遗传突变的功能研究[D]. 南宁:广西医科大学,2020.
[6]Verstraeten VL, Broers JL, van Steensel MA, et al. Compound heterozygosity for mutations in LMNA causes a progeria syndrome without prelamin A accumulation. Hum Mol Genet,2006,15(16):2509-2522.
[7]Wang J, Yu Q, Tang X, et al. Epidemiological characteristics of patients with Hutchinson-Gilford progeria syndrome and progeroid laminopathies in China. Pediatr Res,2024,95(5):1356-1362.
[8]Ullrich NJ, Gordon LB. Hutchinson-Gilford progeria syndrome. Handb Clin Neurol,2015,132:249-264.
[9]余佳,桑艳梅.儿童早老症研究进展.中华实用儿科临床杂志,2021,36(2):3.
[10]Greer MM, Kleinman ME, Gordon LB, et al. Pubertal Progression in Female Adolescents with Progeria. J Pediatr Adolesc Gynecol,2018,31(3):238-241.
[11]Prakash A, Gordon LB, Kleinman ME, et al. Cardiac abnormalities in patients with Hutchinson-Gilford Progeria syndrome. JAMA Cardiol,2018,3(4):326-334.
[12]Schnabel F, Kornak U, Wollnik B. Premature aging disorders: A clinical and genetic compendium. Clin Genet,2021,99(1):3-28.
[13]Shen JJ, Brown CA, Lupski JR, et al. Mandibuloacral dysplasia caused by homozygosity for the R527H mutation in lamin A/C. J Med Genet,2003,40(11):854-857.
[14]阳芳,李乾,刘宝华,等.下颌骨肢端发育不良合并早衰的LMNA基因突变研究.临床皮肤科杂志,2023,52(1):7-10.
[15]Starke S, Meinke P, Camozzi D, et al. Progeroid laminopathy with restrictive dermopathy-like features caused by an isodisomic LMNA mutation p.R435C. Aging (Albany NY),2013,5(6):445-459.
[16]Cabanillas R, Cadiñanos J, Villameytide JA, et al. Néstor-Guillermo progeria syndrome: a novel premature aging condition with early onset and chronic development caused by BANF1 mutations. Am J Med Genet A,2011,155a(11):2617-2625.
[17]Coppedè F. Mutations involved in Premature-Ageing syndromes. Appl Clin Genet,2021,14:279-295.
[18]Gordon LB, Harling-Berg CJ, Rothman FG. Highlights of the 2007 Progeria Research Foundation scientific workshop: progress in translational science. J Gerontol A Biol Sci Med Sci,2008,63(8):777-787.
[19]Suzuki M, Jeng LJB, Chefo S, et al. FDA approval summary for lonafarnib (Zokinvy) for the treatment of Hutchinson-Gilford progeria syndrome and processing-deficient progeroid laminopathies. Genet Med,2023,25(2):100335.
[20]Balmus G, Larrieu D, Barros AC, et al. Targeting of NAT10 enhances healthspan in a mouse model of human accelerated aging syndrome. Nat Commun,2018,9(1):1700.