


151、神经节苷脂贮积症
罕见病诊疗指南(2025年版)
概述 神经节苷脂贮积症(gangliosidosis)为一组罕见的常染色体隐性遗传性溶酶体贮积症,因β-半乳糖苷酶(β-galactosidase)或氨基己糖苷酶(β-hexosaminidase, β-Hex)缺乏导致糖脂类复合物在大脑和其他组织中沉积,引起多系统多器官病变,以神经退行性病变最为突出。根据所缺乏的酶及基因变异不同,神经节苷脂贮积症分为GM1神经节苷脂贮积症和GM2神经节苷脂贮积症。
病因和流行病学
1.GM1神经节苷脂贮积症 GM1神经节苷脂贮积症的发病率为活产婴儿的1/100 000~1/200 000。据报道,巴西的发病率为1/17000,罗马的发病率为1/10000,马耳他群岛的发病率为1/3700。
由GLB1基因变异导致β-半乳糖苷酶缺陷,大量的GM1神经节苷脂在各种组织尤其是神经组织中沉积。导致大脑灰质神经元出现空泡变性以及神经元坏死,大脑白质的髓鞘脱失伴星形胶质细胞和小胶质细胞增生。可以看到神经元凋亡、内质网应激、神经纤维轴浆转运异常、神经纤维-少突胶质细胞相互作用异常。坏死或受损的细胞可以激活小胶质细胞引起炎症反应,这种炎症反应在神经退行性病变的病理生理中起重要作用。
2.GM2神经节苷脂贮积症 由于氨基己糖苷酶缺乏导致GM2神经节苷脂在神经系统沉积所致。根据突变基因的种类分为3类:(1)B型(Tay-Sachs病),编码α亚基的HEXA基因变异导致氨基己糖苷酶A(hexosaminidase A,HexA)缺乏;(2)O型(Sandhoff病),编码β亚基的HEXB基因变异导致HexA及氨基己糖苷酶B(hexosaminidase B,HexB)均缺乏;(3)AB型(GM2激活蛋白酶缺陷型),GM2A基因突变。其中Tay-Sachs病的发病率为活产婴儿的1/201 000~1/222 000,Sandhoff病发病率约为1/422 000。
Hex有2种同工酶,分别为HexA和HexB,前者由1个α亚基和1个β亚基组成,后者由2个β亚基组成。编码α亚基的HEXA基因变异导致HexA缺乏,编码β亚基的HEXB基因突变导致HexA和HexB均缺乏,此外只有HexA能水解GM2,且必须依赖GM2A基因编码的GM2激活蛋白酶。因此HEXA、HEXB、GM2A任一基因突变均可引起相应的酶缺陷,从而使GM2神经节苷脂降解发生障碍而在细胞内堆积。
临床表现
1.GM1神经节苷脂贮积症 根据发病年龄和临床表现,分为三种类型。Laur等回顾性研究法国1998—2019年共61个GM1病人,其中Ⅰ型占67%,Ⅱ型占26%,Ⅲ型占7%。
(1)Ⅰ型(婴儿型):最多见,6个月内起病,病情进展快,伴有肌张力减低及严重的神经系统退行性病变,出现精神运动发育倒退,癫痫发作,对声音敏感,喂养不良。查体发现所有患者出现肌张力减低,84%的患者存在锥体束征。半数患儿眼睛视网膜出现樱桃红斑,部分有角膜薄翳;部分患儿出现肝脾大和心肌病,不同程度的全身性骨骼发育不良,胸、腰椎椎体前下缘鸟嘴样突出,类似黏多糖病样改变,随着病程进展表现越明显。其他表现包括皮肤粗糙增厚、多毛、大片蒙古斑和血管角质瘤、面容粗笨、耳大耳位低、牙龈肥厚伴舌大、牙齿萌出异常、宽掌及手指粗短。病情进展迅速,逐渐出现双目失明、肌张力增高、去大脑强直,多在1~3岁死亡。
(2)Ⅱ型(晚期婴儿型/青少年型):通常在7个月~3岁起病,伴有精神运动发育迟缓,半数以上患者容貌丑陋,骨骼发育障碍,30%病人出现肝脾大、心肌病,少数病人眼底樱桃红斑,病情进展缓慢,预期寿命5~10岁。
(3)Ⅲ型(成年型):起病较晚,3~30岁发病,常表现为进行性加重的椎体外系症状,所有患者出现锥体束征和肌张力异常,伴随步态异常和语言障碍以及骨骼发育障碍,部分患者出现心肌病,寿命相对缩短。
2.GM2神经节苷脂贮积症
根据起病年龄,Sandhoff病的临床表现和临床分型与Tay-Sachs病极其相似。Tay-Sachs病可分为婴儿型、青少年型和成年型三种,临床表现具有高度异质性。青少年型和成年型统称为晚发型。
(1)婴儿型:又称经典型,最常见,早期称为家族性黑矇性痴呆症,患儿出生时均正常,生后3~6个月起病,表现为运动发育落后,出现声音刺激特别敏感,表现为突发惊跳和四肢伸展性阵挛,6~10个月呈现精神运动发育倒退的征象,追光反应差,眼震颤,眼底检查可见樱桃红斑,10个月以后病情迅速进展,自主活动消失,对外界反应淡漠,逐渐出现惊厥、失明、肌强直,但无周围神经受累表现,无面部骨骼改变,1岁以后出现去大脑强直,吞咽困难,频繁惊厥,逐渐演变为无反应的植物人状态,多在发病3~5年死于恶病质。
(2)青少年型:起病年龄2~10岁,最初的症状为步态异常、动作不协调、语言障碍和发育落后等,10岁左右出现肌阵挛和惊厥,随着病情进展,逐渐出现运动功能丧失,肌肉萎缩、失明等,少数可见眼底樱桃红斑,10~15岁时出现去大脑强直状态,数年后死亡。
(3)成年型:起病年龄>18岁,病情进展缓慢,具有明显异质性,主要表现为共济失调、语言障碍、动作不协调、精神心理障碍等,眼底检查一般无樱桃红斑,可存活至成年。
辅助检查
1.GM1神经节苷脂贮积症
(1)酶学检查:白细胞和成纤维细胞中β-半乳糖苷酶活性缺乏是诊断GM1神经节苷脂贮积症的金标准。采用人工底物4-甲基伞形酮-β-半乳糖苷测定患者成纤维细胞β-半乳糖苷酶残余酶活性也有助于GM1神经节苷脂贮积症的诊断,婴儿型患者残余0.07%~1.3%酶活性,青少年型患者为0.3%~4.8%,成年型患者为9%,表型的严重程度与残余酶活性有关。
(2)基因检测:GLB1基因位于3p22.3,含有16个外显子。目前发现了大约290种基因突变,由于分子遗传和临床表现存在异质性,GM1神经节苷脂贮积症基因型与表型的关系还不十分清楚。
2.GM2神经节苷脂贮积症
(1)酶学检测:HexA酶活性和HexA&B酶活性测定是诊断Tay-Sachs病和Sandhoff病的重要依据,可采用外周血白细胞和培养皮肤成纤维细胞进行。其中HexA酶活性缺乏、HexB酶活性正常或增高,诊断Tay-Sachs病;HexA&B酶活性显著减低,HexA酶活性轻度减低,诊断Sandhoff病。
(2)基因检测:Tay-Sachs病由HEXA基因突变导致,HEXA基因位于15q23,含14个外显子,目前发现大约245种HEXA基因突变,包括错义/无义突变、剪接突变、小缺失突变、小插入突变、插入缺失突变和大片段缺失突变。Sandhoff病由HEXB基因纯合或复合杂合突变导致,HEXB基因位于5q13.3,含14个外显子,编码556个氨基酸,报道了166种基因突变,其中最常见的突变为一个包含启动子和1~5外显子在内的16kb大片段缺失。
诊断
神经节苷脂贮积症需结合临床表现,对于有精神运动发育落后或倒退,语言障碍,共济失调,椎体外系症状,锥体束征阳性,病情迅速或缓慢进展,伴或不伴面容粗笨、骨骼发育异常、肝脾大、心肌病、眼底樱桃红斑等症状的患者,进一步行酶学及基因学检查明确诊断。
鉴别诊断
神经元蜡样质脂褐质沉积症:本病表现为频发的肌阵挛发作,视觉改变,视网膜色素变性,共济失调,进行性痴呆等,确诊需要皮肤或脑组织活组织检查在电镜下发现特异性的包涵体(膜性颗粒物质、曲线体和指纹体样结构)。
治疗
神经节苷脂贮积症呈进行性进展的病程,早期诊断、早期治疗可减缓底物积聚,从而改善临床和预后。目前针对GM1、GM2神经节苷脂贮积症没有特效疗法,只有对症和支持治疗。其他治疗策略包括骨髓移植治疗、基因治疗、底物减少、分子伴侣治疗等,目前尚处于研究阶段。
遗传咨询
神经节苷脂贮积病是常染色体隐性遗传病,患儿父母再次怀孕应进行。
诊疗流程(图30-1, 30-2)
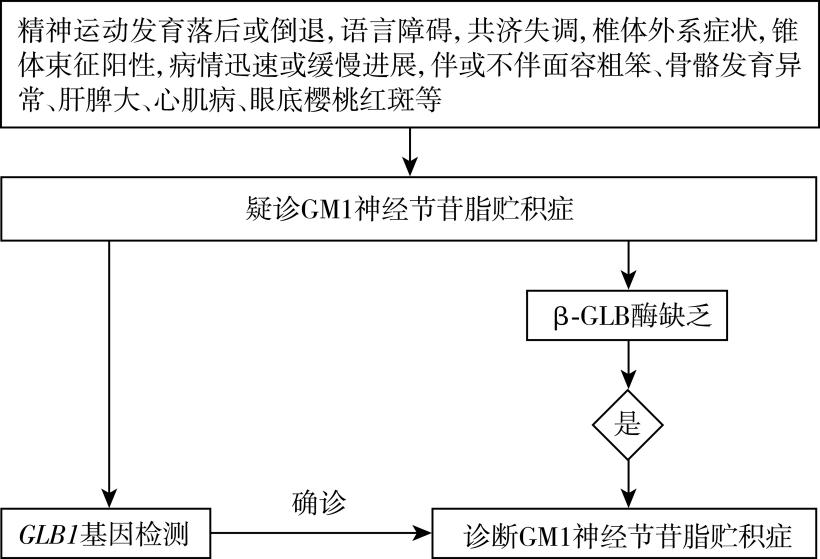
图30-1 GM1神经节苷脂贮积症诊疗流程
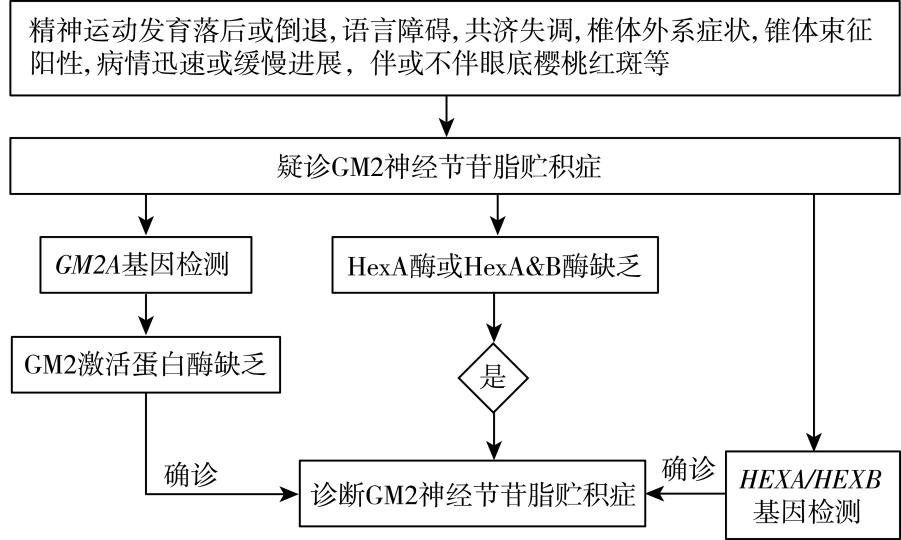 图30-2 GM2神经节苷脂贮积症诊疗流程
图30-2 GM2神经节苷脂贮积症诊疗流程
参考文献
[1]Parenti G, Medina DL, Ballabio A. The rapidly evolving view of lysosomal storage diseases. EMBO Mol Med,2021,13(2):e12836.
[2]Parenti G, Andria G, Ballabio A. Lysosomal storage disease: from pathophysiology to therapy. Annu Rev Med,2015,66(1):471-486.
[3]Ferreira CR, Gahl WA. Lysosomal storage diseases. Transl Sci Rare Dis,2017,2(1-2):1-71.
[4]Brunetti-Pierri N, Scaglia F. GM1 gangliosidosis: a review of clinical, molecular, and therapeutic aspects. Mol Genet Metab,2008,94(4):391-396.
[5]Sinigerska I, Chandler D, Vaghjiani V, et al. Founder mutation causing infantile GM1-gangliosidosis in the gypsy population. Mol Genet Metab,2006,88(1):93-95.
[6]Meikle PJ, Hopwood JJ, Clague AE, et al. Prevalence of lysosomal storage disorders. JAMA,1999,281(3):249-254.
[7]Sandhoff K, Andreae U, Jatzkewitz H. Deficient hexozaminidase activity in an exceptional case of Tay-Sachs disease with additional storage of kidney globoside in visceral organs. Life Sci,1968,7(6):283-288.
[8]Regier DS, Tifft CJ, Rothermel CE. GLB1-related disorders//ADAM MP, FELDMAN J, MIRZAA GM, et al. eds. GeneReviews®. Seattle (WA): University of Washington,2013.
[9]Jarnes Utz JR, Kim S, King K, et al. Infantile gangliosidoses: mapping a timeline of clinical changes. Mol Genet Metab,2017,121(2):170-179.
[10]Laur D, Pichard S, Bekri S, et al. Natural history of GM1 gangliosidosis-retrospective cohort study of 61 French patients from 1998 to 2019. J Inherit Metab Dis,2023,46(5):972-981.
[11]Bley AE, Giannikopoulos OA, Hayden D, et al. Natural history of infantile G(M2) gangliosidosis. Pediatrics,2011,128(5):1233-1241.
[12]Maegawa GH, Stockley T, Tropak M, et al. The natural history of juvenile or subacute GM2 gangliosidosis: 21 new cases and literature review of 134 previously reported. Pediatrics,2006,118(5):1550-1562.
[13]Federico A, Palmeri S, Malandrini A, et al. The clinical aspects of adult hexosaminidase deficiencies. Dev Neurosci,1991,13(4-5):280-287.
[14]Neote K, McInnes B, Mahuran DJ, et al. Structure and distribution of an Alu-type deletion mutation in Sandhoff disease. J Clin Invest,1990,86(5):1524-1531.
[15]梁雁,罗小平.提高对溶酶体贮积症诊断与治疗的认识.中华儿科杂志,2021,59(6):435-438.