


149、进行性骨化性纤维发育不良
罕见病诊疗指南(2025年版)
概述 进行性骨化性纤维发育不良(fibrodysplasia ossificans progressiva, FOP),又称为进行性骨化性肌炎(myositis ossificans progressiva, MOP)或“石人病(stone man disease)”,是由于ACVR1/ALK2基因突变引起的一种罕见的具有严重致残性的遗传性结缔组织疾病,其典型特征是踇趾先天性畸形和进行性异位软骨内骨化,关节受累并导致异位骨化,活动能力降低,并导致严重的残疾。
病因和流行病学
FOP是由染色体2q24上的ACVR1/ALK2基因突变引起BMP/TGFβ信号通路失调导致,无论是散发性还是家族遗传性FOP,均存在ACVR1基因的杂合子错义突变,绝大多数患者的突变位点是c.617G>A (p.R206H)。少部分患者也存在其他类型的突变,例如在中国患者中确认存在的突变位点:c.774G>C (p.R258S)和c.1067G>A (p.G356D)。ACVR1/ALK2是TGF-β受体家族中的一种骨形态发生蛋白(BMP) Ⅰ型受体,是BMP信号途径的一部分,该途径在正常的骨发育和修复中起到关键作用。ACVR1蛋白含有胞外N端配体结合结构域、跨膜(TM)结构域、甘氨酸-丝氨酸(GS)结构域和蛋白激酶(PK)结构域。最常见的FOP相关ACVR1基因突变导致ACVR1蛋白的GS结构域第206位氨基酸由精氨酸变为组氨酸。GS结构域是ACVR1与FKBP12的结合位点,也是激活特异性Smad信号蛋白的关键位点。FKBP12是一种抑制蛋白,在没有配体的情况下,它与GS结构域结合,可以抑制其酶活性并防止ACVR1激活。GS结构域的突变,使ACVR1与FKBP12的结合力减弱,降低FKBP12的抑制作用,导致ACVR1-BMP信号传导激活。此外,GS结构域突变还可能导致ACVR1对各种BMP配体和激活素A具有异常反应,通过Smad1/5/8信号蛋白进一步增强BMP信号转导。过度活跃的BMP信号导致FOP的异位骨化。
FOP的患病率约为1/120万~1/200万,且无性别、种族、民族或地理易感性,目前尚无已知的危险因素。大多数FOP是散发的,但也有一些家族性FOP病例呈现常染色体显性遗传规律。目前在我国报道的FOP患者均为散发性基因突变。
临床表现
经典型FOP临床表现主要为踇外翻畸形与进行性异位骨化(HO)。除此之外,大于50%的FOP患者有胫骨内侧近端骨软骨瘤、颈椎原位融合、股骨颈短而宽、传导性听力障碍和踇指畸形。
FOP患者在出生时表现正常,仅出现特征性踇趾畸形,其特征是踇趾短和踇外翻,第一跖骨畸形,第一趾指间关节缺失或融合。大多数患者在10岁之内会出现不同程度的急性软组织肿胀、疼痛,常见于后颈部和背部,一般不需要格外处理,肿胀可以自行消退,但更常见的是逐渐恶化并最终发展为成熟的异位骨。异位骨化多发生于肌腱、韧带、筋膜和骨骼肌等处,其特点为从头部、颈部和肩部开始,从颅骨到尾骨、背侧到腹侧、轴向到尾端、近端到远端进展。膈肌、舌肌、眼外肌、心肌和平滑肌不受影响。软组织损伤、肌内注射、手术切口、跌倒、肌肉拉伸或病毒性疾病都可能导致急性发作。
异位骨化逐渐积累,最终延伸到关节会导致进行性和不可逆的活动困难,大多数患者在7岁左右出现异位软骨内成骨, 15岁左右出现脊柱和上肢活动严重受限,大多数FOP患者30岁左右便无法独立生活并需要依靠轮椅。FOP患者异位骨化累及颈椎椎体导致跨节段的桥接骨形成,引起颈强直。累及颞下颌关节会导致吞咽困难并出现体重减轻。累及中耳耳骨出现传导性的听力障碍,常见于儿童期或青春期患者。累及肋间肌、肋椎关节和胸椎旁软组织会导致胸廓功能不全综合征(thoracic insufficiency syndrome,TIS),导致呼吸困难,严重危害FOP患者生命安全。大多数患者死于TIS相关并发症,如肺炎和心脏功能衰竭。
辅助检查
1.常规生化检查(如血清钙、磷、碱性磷酸酶、甲状旁腺激素水平、肾功能、尿钙、磷酸盐) 通常正常。在异位骨化形成的急性发作期间,一些患者的碱性磷酸酶(ALP)和红细胞沉降率(ESR)可能升高。
2.影像学 FOP的影像学特征包括关节畸形,尤其是踇趾(如双侧踇外翻畸形、第一跖骨畸形、指间关节缺失或融合)和软组织骨化。除此之外,FOP的各种X线表现还包括胫骨内侧近端骨软骨瘤(约90%),颈椎后路植骨融合(约80%),股骨颈宽、短(约50%),以及拇指畸形(大约50%)。
3.病理 FOP病理特点分为三个阶段,早期镜下示组织结构破坏伴单核细胞、巨噬细胞、肥大细胞和T淋巴细胞浸润,该阶段为分解代谢阶段;随后进入合成代谢阶段,即大量新生毛细血管及纤维组织增生,增生的纤维组织逐渐过渡成软骨;最后血运重建和成骨,异位骨是成熟的板层骨组织,可含骨髓。
4.分子遗传学 主要为ACVR1/ALK2基因蛋白编码区的错义突变或框内缺失。
诊断
FOP是一种经分子遗传学证实的临床诊断。儿童出现先天性踇趾畸形、进行性软组织肿胀和头部或背部结节应考虑FOP的可能性。对于高度怀疑FOP的患者,在完善影像学检查的基础上,通过基因检测明确诊断。软组织病变的穿刺活检可能会引起急性发作,应尽量避免。
鉴别诊断
常见的需要与FOP鉴别诊断的疾病包括:
1.皮肌炎 可能表现为肌肉疼痛、无力和肿胀。这些症状可能与FOP早期相似。血清中肌酸激酶(CK)和乳酸脱氢酶(LDH)水平增高有助于诊断。
2.骨化性肌炎(myositis ossificans) 通常是非遗传性的,与FOP不同,多发生在大肌群,如大腿和臂部,常常是由于外伤或其他未知原因导致。其影像学特征和病程与FOP有所不同,FOP的骨化多发生在身体的中轴线,而骨化性肌炎则更多的是在单一的肌肉或肌肉群受累。
3.慢性复发性多灶性骨髓炎(chronic recurrent multifocal osteomyelitis, CRMO) 表现为反复发作的骨髓炎,可能伴有疼痛、红肿和发热等症状。通过骨生物学检查、影像学及其他相关实验室检查可以与FOP鉴别。
4.肌纤维母细胞瘤 这是一种良性的软组织肿瘤,起源于肌肉的结缔组织。它可能会形成肿块并导致局部疼痛或不适,与FOP早期的肿块形成类似,但无先天性踇趾畸形,通过细胞病理学检查和影像学检查可以进行鉴别。
5.进行性骨化性异位增生症(progressive osseous heteroplasia,POH) 是由于GNAS基因突变导致的一种罕见遗传性疾病,以皮肤骨化为特征,进展到皮下和深部结缔组织。虽然POH获得性异位骨化也发生在创伤部位,但在幼儿中很少见,通常无踇趾畸形或快速进行性软组织肿胀。
治疗
FOP的管理主要为支持性,着重于预防突发事件、对患者及家庭进行教育、咨询,以及提高生活质量。
1.预防
(1)避免伤害:任何伤害或创伤,即使是微小的,都可能触发新的骨化。因此,避免活检、手术、注射、牙科手术或其他可能伤害软组织的活动,并努力预防跌倒十分重要。
(2)疫苗接种:接种灭活流感疫苗以降低流感引起的FOP发作,应避免减毒流感活疫苗,选择皮下接种,并避免在受FOP影响的关节或肌肉附件接种。疫苗应避免在FOP发作期或病发后的6~8周内接种。可皮下接种的疫苗包括MMR、水痘和脊髓灰质炎病毒灭活疫苗。应避免含有白喉或破伤风类毒素的疫苗(例如,白喉、破伤风和百日咳的组合疫苗),因其可能导致突发、异位骨化和关节活动永久丧失。
2.症状缓解与管理
(1)非甾体抗炎药(NSAIDs):对于急性炎症期的FOP,NSAIDs(如布洛芬)可用于减少炎症和疼痛。
(2)糖皮质激素:对于影响颌骨、颌下区域或主要关节(如髋关节)的病变,需要短程的糖皮质激素治疗。泼尼松1~2mg/kg,每天口服一次或两次(每日最大剂量100mg),持续3~4天。也可以给予同等剂量的其他口服糖皮质激素。糖皮质激素通常不用于颈部或躯干的治疗,因为这些部位的发作通常是长期和反复的。停用糖皮质激素后,可在发作的剩余时间内使用局部或全身非甾体抗炎药物(如布洛芬)或局部应用冷敷对症治疗。此外,在严重软组织创伤或手术(如阑尾切除术、牙科手术)后24小时内使用短疗程的糖皮质激素可以防止急性发作。
3.其他药物选择
(1)帕罗伐汀(palovarotene):一种选择性的视黄醇受体γ激动剂,已经在临床试验中显示出抑制FOP异位骨化的潜力。
(2)REGN-2477:一种激活素A抗体,在美国和欧洲已经显示出良好的临床前结果,目前仍处于临床试验阶段。
(3)西罗莫司:一种mTORC1抑制剂,是一种常用的免疫抑制剂,目前正在日本进行临床试验。
(4)抗白介素药物:抗白介素药物(anakinra和canakinumab)似乎有助于降低急性发作的频率,但抗白介素药物治疗仍在研究中。
4.综合管理
(1)预防性牙科护理:是FOP管理的重要组成部分,应该尽快开始,建议在进行任何牙科手术前咨询具有FOP专业知识的牙科专业人士,牙科手术应避免注射局部麻醉剂。
(2)听力评估:患有FOP的儿童出现传导性听力丧失的风险增加,应至少每2年接受一次听力测试评估听力损伤。
(3)呼吸健康:FOP患者的胸部畸形和脊柱侧凸可能会降低呼吸能力,可以通过积极的呼吸活动(例如,歌唱、激励性肺活量测定、峰值流速哨声)来维持呼吸健康。
(4)心理和社会支持:由于FOP的严重性和进展性,患者和家庭可能需要心理和社会支持来应对与疾病相关的心理和情感压力。
诊疗流程(图28-1)
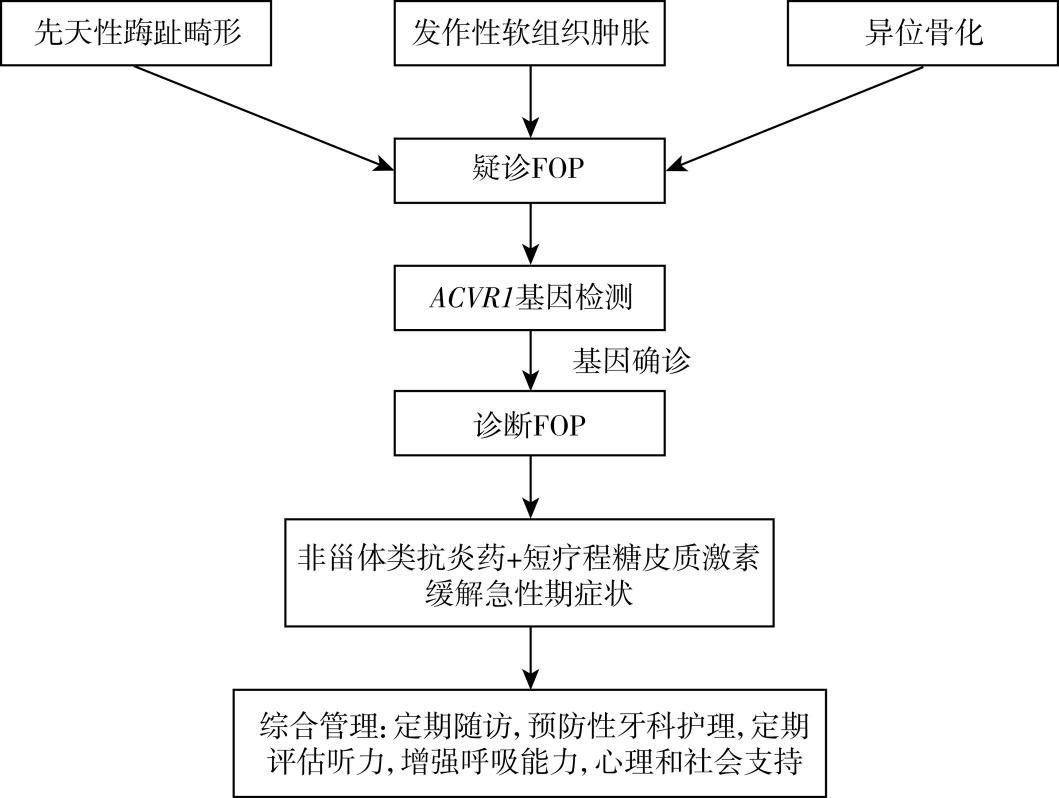
图28-1 进行性骨化性纤维发育不良的诊疗流程
参考文献
[1]PIGNOLO RJ, SHORE EM, KAPLAN FS. Fibrodysplasia ossificans progressiva: diagnosis, management, and therapeutic horizons. Pediatr Endocrinol Rev,2013,10 Suppl 2(02):437-448.
[2]SHE D, ZHANG K. Fibrodysplasia ossificans progressiva in China. Bone,2018,109:101-103.
[3]KAPLAN FS, AL MUKADDAM M, STANLEY A, et al. Fibrodysplasia ossificans progressiva (FOP): a disorder of osteochondrogenesis. Bone,2020,140:115539.
[4]KALIYA-PERUMAL AK, CARNEY TJ, INGHAM PW. Fibrodysplasia ossificans progressiva: current concepts from bench to bedside. Dis Model Mech,2020,13(9):dmm046441.
[5]KAPLAN FS, XU M, GLASER DL, et al. Early diagnosis of fibrodysplasia ossificans progressiva. Pediatrics,2008,121(5):e1295-e1300.
[6]TOWLER OW, SHORE EM, KAPLAN FS. Skeletal malformations and developmental arthropathy in individuals who have fibrodysplasia ossificans progressiva. Bone,2020,130:115116.
[7]ALESSI WOLKEN DM, IDONE V, HATSELL SJ, et al. The obligatory role of activin A in the formation of heterotopic bone in fibrodysplasia ossificans progressiva. Bone,2018,109:210-217.
[8]AYKUL S, HUANG L, WANG L, et al. Anti-ACVR1 antibodies exacerbate heterotopic ossification in fibrodysplasia ossificans progressiva (FOP) by activating FOP-mutant ACVR1. J Clin Invest,2022,132(12):e153792.