


134.慢性炎性脱髓鞘性多发性神经根神经病
罕见病诊疗指南(2025年版)
概述 慢性炎性脱髓鞘性多发性神经根神经病(chronic inflammatory demyelinating polyradiculoneuropathy,CIDP)是一类由免疫介导的脱髓鞘性周围神经病,病情进展达8周以上,可有缓解复发过程;电生理表现为周围神经传导速度减慢、远端潜伏期延长、运动神经传导阻滞、异常波形离散以及F波异常等脱髓鞘改变,大部分患者免疫治疗有效。
病因和流行病学
CIDP发病为细胞和体液免疫共同参与结果,临床有多种亚型,不同亚型的具体发病机制可能有所不同。CIDP患病率(0.67~10.3)/10万,年发病率(0.15~10.6)/10万。各个年龄均可发病,高峰期40~60岁,也有儿童甚至新生儿发病的报道,男性略多于女性。
临床表现
按照周围神经病变的分布特点和受累纤维种类,可以将CIDP分为两个大类,即CIDP经典型和CIDP变异型。经典型CIDP临床最为常见,表现为多发性周围神经病,对称性上下肢近端和远端无力,并伴有肢体远端的感觉异常,下肢重于上肢。变异型CIDP包括五种亚型:①远端型CIDP:主要表现为肢体远端为主的感觉运动型周围神经病,下肢重于上肢,肢体近端肌力通常正常或仅有轻微受累;②多灶型CIDP:符合多发单神经病的特点,感觉异常和无力以多灶的模式不对称分布,有一个以上肢体受累,通常以上肢为主,部分患者可有脑神经受累;③局灶型CIDP:感觉和运动受累局限于一个肢体,症状类似于臂丛或腰骶神经丛病变;④纯运动型CIDP:符合多发性周围神经病分布,下肢受累重于上肢,只有运动受累的表现,而无感觉异常;⑤纯感觉型CIDP:符合多发性感觉性周围神经病,下肢重于上肢,仅有感觉受累的症状体征,无运动受累。在经典型和变异型CIDP表现为多发性周围神经病者,所有肢体的腱反射均减低或消失;在局灶型或多灶型CIDP,不受累肢体的腱反射可以正常。随着病情进展,部分CIDP变异型可能发展为典型的CIDP表现。
无论是经典型还是变异型CIDP,病程特点并无差异,均可表现为进行性发展或有复发缓解的过程,病程发展至少达8周。CIDP病程发展是指自起病至达到病情最重的时间,不等同于发病至就诊的时间。部分CIDP患者起病较急,早期可类似吉兰-巴雷综合征,但随诊观察时可见超过4周病情仍进展,或治疗好转后,停药复发,通常复发次数大于2次,应考虑为CIDP。
辅助检查
1.肌电图 CIDP是一种脱髓鞘性周围神经病,肌电图是诊断CIDP最重要的辅助方法,是确定CIDP诊断的必需条件,主要用于提供脱髓鞘病变的证据。脱髓鞘的证据主要依靠运动神经传导、F波和感觉神经传导测定。判断周围神经脱髓鞘时,主要依据运动神经传导测定,电生理参数包括:运动远端潜伏期延长、运动传导速度下降、F潜伏期延长/F波出现率下降、运动传导阻滞、异常波形离散以及远端复合肌肉动作电位(compound muscle action potential, CMAP)负向波时限增宽。CIDP的感觉传导异常可表现感觉传导速度下降,感觉神经动作电位波幅下降,在CIDP可以出现正中神经或桡神经感觉传导异常,而腓肠神经感觉传导正常的现象。除了纯感觉型CIDP外,在诊断其他类型CIDP时,均要求至少在两根运动神经存在肯定脱髓鞘病变的证据;除了纯运动型CIDP外,至少应有两根感觉神经存在传导异常。诊断感觉型CIDP时,至少要求两根神经在非嵌压部位感觉传导速度明显减慢,而运动传导完全正常。
2.脑脊液检测 CIDP患者脑脊液检查可见白细胞数正常范围,蛋白增高,即蛋白细胞分离现象。在部分CIDP患者,脑脊液蛋白可以正常。如果临床和电生理检测能够诊断CIDP,可不必进行脑脊液检测。在下述情况时,可以考虑进行脑脊液检测来辅助CIDP诊断和鉴别诊断:当电生理检测仅在一根神经发现脱髓鞘证据时,脑脊液蛋白细胞分离可以作为诊断的支持依据;当患者急性或亚急性起病,怀疑有感染或恶性肿瘤,应行脑脊液检测协助鉴别诊断。蛋白细胞分离现象并非CIDP特异性改变,可见于多种临床情况。当电生理检查缺乏明确的脱髓鞘证据时,仅仅有脑脊液蛋白的增高,诊断CIDP需慎重,如腰椎管狭窄患者,可有蛋白增高,糖尿病或其他原因导致的周围神经病也可见蛋白增高现象,常会误诊为CIDP,而进行不必要的免疫治疗。在50岁以上患者,脑脊液正常值较年轻的患者增高,目前尚缺乏诊断CIDP的蛋白升高的界值。腰穿的另一个主要价值在于鉴别其他疾病,如是否存在感染或恶性肿瘤浸润导致的白细胞增高。
3.影像学表现 CIDP的周围神经超声可表现为周围神经横截面积增粗,神经束信号异常,如果在非嵌压部位,出现周围神经节段性的明显增粗,更有支持价值;同一患者不同神经的表现可有明显差异,可见正常神经与增粗神经并存。少数患者周围神经超声检测神经横截面积可均在正常范围。CIDP周围神经形态的改变特点,可能与检测时患者的病程、病情严重程度、治疗与否、潜在发病机制有关。在CIDP患者磁共振检测可见神经根有增粗,或T2序列高信号,神经根或神经干的定量检测或半定量评分较定性判断更有价值。当电生理检测证实至少2根神经存在脱髓鞘证据时,不必进行周围神经超声或磁共振检测;当电生理检测仅发现一根神经存在脱髓鞘证据时,如果在正中神经、尺神经或臂丛,至少两个部位有神经增粗,可支持CIDP的诊断。
4.腓肠神经病理检查 CIDP主要病理改变为有髓神经纤维出现节段性脱髓鞘,轴索变性,雪旺氏细胞增生并形成洋葱球样结构,单核细胞浸润等。在CIDP的诊断过程中,不建议常规进行周围神经活检,只有在某些特定临床情况下,有必要时,根据情况选择,比如,在高度疑诊CIDP的患者,临床、电生理、影像学和脑脊液检查均仍不能确定诊断时;或疑诊CIDP,但免疫治疗无效,或还需要与其他疾病鉴别,如遗传性运动感觉神经病、淀粉样变性、血管炎、结节病、神经鞘瘤/神经纤维瘤等。
5.其他化验 CIDP缺乏特异性的诊断指标,化验检查一方面主要用于鉴别其他疾病,另一方面为免疫治疗方案的选择和监测不良反应提供依据。
(1)单克隆球蛋白(M蛋白)检测:对于临床疑诊CIDP的成年患者应常规进行M蛋白检测,包括血清蛋白电泳和免疫固定电泳,尿免疫固定电泳以及轻链的检测。在远端型CIDP,如果检测到IgM型M蛋白,应进一步检测抗髓鞘相关糖蛋白(myelin-associated glycoprotein,MAG)抗体,如果二者均阴性,可考虑重复检测。由于M蛋白在50岁以上健康人群可达1%~3.2%,70岁以上人群可达5%,不同类型的M蛋白相关周围神经病的表现不同,因此在发现M蛋白阳性时,要注意识别其与周围神经病究竟是伴随关系还是因果关系。
(2)郎飞氏结和结旁抗体检测:对于临床疑诊CIDP的患者,应考虑抗郎飞氏结和结旁抗体检测。对于存在以下特点者,建议进行相关抗体的检测:IVIG和糖皮质激素治疗效果不佳者;急性或亚急性起病、曾诊断为GBS或急性起病的CIDP(acute onset-CIDP,A-CIDP)者;有低频震颤、明显共济失调或明显远端为主无力者;呼吸衰竭和有脑神经受累者;有肾病综合征者;脑脊液蛋白特别高者。抗NF155、抗CNTN1、抗Caspr1以及抗NF140/186抗体的检测,应选择标准化的检测方法,并保证其质量控制。
(3)其他化验检查与鉴别诊断:多种周围神经病在电生理检查时可见明确或可疑的髓鞘病变,需要与CIDP鉴别。根据临床具体情况,可选择不同的检测项目,如血沉,C反应蛋白,抗核抗体、抗中性粒细胞胞浆抗体、抗神经节苷脂抗体、肌酶、维生素B12和B6、副肿瘤抗体、Lyme抗体、HIV、骨穿、胸部X线,基因检测等。治疗前应常规进行血尿常规、肝肾功能和血糖、骨密度等,用于监测治疗药物的不良反应。
诊断
1.周围神经病变自发生至达到高峰的时间至少8周,可表现为慢性进展或缓解复发。
2.符合CIDP临床表现,并且电生理证实至少在两根神经存在脱髓鞘的证据,并排除其他原因导致的脱髓鞘周围神经病。
3.符合CIDP临床表现,但电生理证实仅有一根神经存在脱髓鞘证据时,如果免疫治疗(IVIg、血浆置换或糖皮质激素)有效,周围神经影像学发现神经明显增粗,脑脊液蛋白细胞分离,神经活检,四项中有一项符合CIDP表现,并排除其他原因导致的脱髓鞘性周围神经病,可诊断CIDP。
鉴别诊断
多种其他疾病也可以出现髓鞘病变,如遗传压迫易感性周围神经病、Charcot-Mari-Tooth1型、POEMS综合征等,因此在电生理符合脱髓鞘诊断的情况时,在临床实践中,仍需结合临床进行鉴别诊断。
1.吉兰-巴雷综合征 A-CIDP在早期通常难以与GBS鉴别,有13%的CIDP患者4周内可类似GBS,5%的GBS患者后来诊断为CIDP。如果IVIg治疗后好转,但复发3次或以上的GBS,则应为CIDP,而非复发性GBS。与GBS不同,CIDP脑神经、呼吸肌、自主神经通常不受累。尽管有上述鉴别线索,在实际的临床工作中,发病4周内,其实很难准确地识别出究竟是GBS,还是A-CIDP,临床通常会采用IVIg治疗。A-CIDP患者,在IVIg治疗1~2个月后,通常会再次加重。在临床诊断的某些GBS患者,由于经济因素影响,无法接受IVIg治疗,而给予了糖皮质激素,临床也有好转,可能与这一组疾病有关。
2.M蛋白血症相关周围神经病 POEMS综合征早期,临床、电生理和脑脊液表现均可类似CIDP,部分患者早期糖皮质激素治疗可略有改善,但维持治疗时,会出现复发持续进展。M蛋白检测有助于二者的识别。当出现皮肤变黑、肢体明显水肿时,可提示POEMS综合征,POEMS综合征的电生理改变传导速度减慢较为均一,近端为主,通常无传导阻滞和异常波形离散,周围神经超声神经增粗程度轻微,通常无局灶性神经增粗。在远端型CIDP患者中,约2/3伴有IgM型单克隆球蛋白,其中部分患者有抗MAG抗体阳性,而MAG抗体阳性者大部分患者发病机制、病理和治疗反应等多个方面均与CIDP不同。
3.抗郎飞氏结和结旁抗体相关周围神经病 也称免疫性郎飞氏结病,其临床、电生理和脑脊液改变与CIDP相似,但可血清中检测到抗CNTN1抗体、NF155抗体、Caspr1抗体或NF140/186抗体。不同抗体的临床表现有所不同,电生理检查通常可以达到CIDP的脱髓鞘病变诊断标准,以前一直归类为CIDP的亚型,在早期研究中,这组患者也通常是从临床表现为CIDP的难治性患者中通过抗体检测而筛查出来。但目前认为,这组疾病不同于CIDP,不同抗体相关的疾病各有其相对独特的临床表现,病理学上均无明显的炎症或巨噬细胞介导的脱髓鞘病变,对IVIg反应差,因此将其从CIDP中分离出来,作为相对独立的一组疾病。该组疾病可选择糖皮质激素治疗,如效果不佳,可考虑利妥昔单抗治疗。
4.遗传性周围神经病 如CMT1A,CMTX,CMT4C等,临床均表现为慢性感觉运动性周围神经病,临床进展非常缓慢,脑脊液可有蛋白细胞分离现象,神经传导测定可见传导速度减慢、远端潜伏期延长等髓鞘病变的特点,但一般无传导阻滞和明显的异常波形离散,患者常有家族史、弓形足等提示遗传性疾病的线索。另外在脑白质营养不良相关疾病、遗传代谢性疾病中,也有少数以周围神经病变为主要表现者,电生理表现为髓鞘病变的特点,需要注意鉴别。
5.中毒性周围神经病 如正己烷中毒,某些生物制剂也可出现类似CIDP的不良反应,如免疫检查点抑制剂。临床表现为多发性感觉运动性周围神经病,电生理表现为脱髓鞘改变的特点,脑脊液可有蛋白细胞分离现象,早期可误诊为吉兰-巴雷综合征,随病情迁延发展,常需要与CIDP进行鉴别,发病前用药史以及流行病学史对于诊断最为重要。
6.系统性疾病伴随脱髓鞘性周围神经病 副肿瘤综合征、淋巴瘤相关周围神经病、HIV感染、少数结缔组织病等均可出现类似CIDP的表现,可根据患者具体情况进行筛查鉴别。
7.多灶性运动神经病 为纯运动型周围神经病,早期为多发单神经病,随病情进展,可呈现类似多发性周围神经病表现。电生理检测可见传导阻滞,需要与CIDP变异型中的多灶性、局灶性以及纯运动型CIDP鉴别。前二者均有感觉纤维的受累,纯运动型CIDP则下肢起病多见,相对对称起病。MMN采用IVIg治疗有效,但对糖皮质激素治疗反应不佳,甚至加重。
8.其他 在局灶性或多灶性CIDP还需要与多种其他原因导致的臂丛或腰骶丛病变、多发单神经病鉴别,如周围神经肿瘤、压迫易感性周围神经病、淀粉样变性周围神经病、结缔组织病相关周围神经病等。纯感觉性CIDP需要与多种药物、副肿瘤综合征、结缔组织病等相关周围神经病鉴别。
治疗
CIDP的治疗包括免疫治疗、对症治疗、营养神经治疗和康复治疗。
1.免疫治疗药物 CIDP的一线治疗包括糖皮质激素和IVIg,血浆置换也是一线治疗方案之一。在长期维持治疗时,前二者是更为优先的选择。血浆置换主要用于糖皮质激素和IVIg效果不佳或病情急性加重时。临床上大约80%的患者一线治疗药物有效。运动型CIDP对于激素反应通常较差,部分患者甚至病情加重,而IVIg治疗有效。
(1)糖皮质激素治疗方案:有多种,临床以口服方案最为常用,泼尼松1mg/(kg·d)或每日60mg开始,维持4周,之后缓慢逐渐减量,根据临床好转情况,每2~4周减5~10mg。糖皮质激素治疗效果的判断通常需要3个月的时间。治疗有效时,可在6个月左右减量至每日20mg或以下维持,在病情稳定的患者,早期每半年或1年评估是否可以进一步减量或停药,长期维持者可1~2年评估一次。在病情较重者,可以选择甲泼尼龙冲击治疗,如甲基强的松龙500~1000 mg/d i.v.gtt.,连续3~5天之后改为口服维持;也有选择定期甲泼尼龙冲击治疗方案,目前并无充分证据表明哪一种方案更有效或副作用更小。治疗过程中,注意补钾补钙保护胃黏膜治疗,定期检查血常规、肝肾功能、血糖、血压,早期每1~2周一次,长期维持者可3~6个月一次,骨密度检测可6个月一次。
(2) IVIg治疗方案:400mg/(kg·d),每天1次,静脉滴注,连续3~5天。通常在用药3~4周时达到最佳效果,之后需要定期维持。目前尚缺乏统一的维持方案,可根据患者用药后无力的变化,个体化定期使用,如每1~2个月输注一次,每次3~5天。IVIg的效果通常在使用后3周之内即可看到效果。如果第一次IVIg治疗无效,可再次给予足量治疗一次,如仍无效,可判断无效,在15%的患者第二次治疗后可有缓解。
(3)血浆置换(plasma exchange,PE)治疗方案:每次血浆交换量为30~50ml/kg,在1~2周内进行3~5次。根据情况可间隔1~2个月定期使用。需要注意的是,在应用IVIg后3周内,不宜进行PE治疗。PE的禁忌证主要是严重感染、心律失常、心功能不全、凝血系统疾病等;其副作用为血流动力学改变,可能造成血压变化、心律失常,使用中心导管可引发气胸和出血以及可能合并败血症。
如一线治疗无效、激素无法耐受,或激素依赖(如泼尼松减量至每日20mg时即复发)等情况时,可考虑使用免疫抑制剂,常用药物包括硫唑嘌呤、吗替麦考酚酯和环孢素。硫唑嘌呤用法为2~3mg/(kg·d),分2~3次口服;环孢素:3~6mg/(kg·d),分2~3次口服。吗替麦考酚酯:2~3g/d,分2~3次口服。有研究显示,硫嘌呤甲基转移酶活性或基因检测可预测其骨髓和肝脏毒性害,指导硫唑嘌呤的使用,避免严重不良反应。
当这三个药物无效时,可考虑使用环磷酰胺或利妥昔单抗。环磷酰胺:可200~400mg每2周一次,静脉注射,或口服50mg每日两次。利妥昔单抗100~600mg,每周一次,静脉输液,共4周,半年后根据情况再用。
尽管免疫抑制剂治疗仍缺乏充分的循证依据,但部分研究仍提示可以选择为减少一线药物治疗剂量的辅助用药。目前尚缺乏生物学指标,用于指导不同免疫抑制剂的选择,也无法判断哪一种免疫抑制剂对某一患者更为有效或无效。
目前有关于CIDP的多项临床试验正在开展,效果有待证实。艾加莫德皮下注射制剂治疗CIDP的临床试验显示,该药可改善CIDP的多项临床功能评分,维持治疗可降低CIDP复发风险,已在部分国家被批准用于CIDP的治疗。
不推荐甲氨蝶呤、β-1a干扰素、芬戈莫德用于CIDP的治疗,在获得更多临床证据前,不建议常规使用他克莫司、α干扰素,以及其他单抗类,如阿仑单抗、那他珠单抗,以及硼替佐米、益赛普、氨基吡啶、氟达拉滨、阿巴西普、免疫吸附。
免疫药物的选择和维持方案,需要注意个体化。如对于有生育需求、妊娠或哺乳期的女性CIDP患者,慎用免疫抑制剂,因病情需要必需使用时,可以选择硫唑嘌呤、环孢素、糖皮质激素和IVIg。吗替麦考酚酯、环磷酰胺和利妥昔单抗均增加致畸风险,应至少停药6个月再怀孕。
2.对症治疗及神经营养治疗 少数CIDP患者可有神经痛,可使用加巴喷丁、普瑞巴林、阿米替林等。尽管缺乏循证证据,临床常给予VitB1、复合维生素B和B12等营养神经治疗。
3.功能锻炼及康复 运动和感觉受累所导致的残疾,可以通过功能训练逐渐恢复,足部支具有助于改善患者行走的稳定性和步态,健康积极的生活态度和生活方式等有益于CIDP患者功能的恢复。
诊疗流程(图13-1)
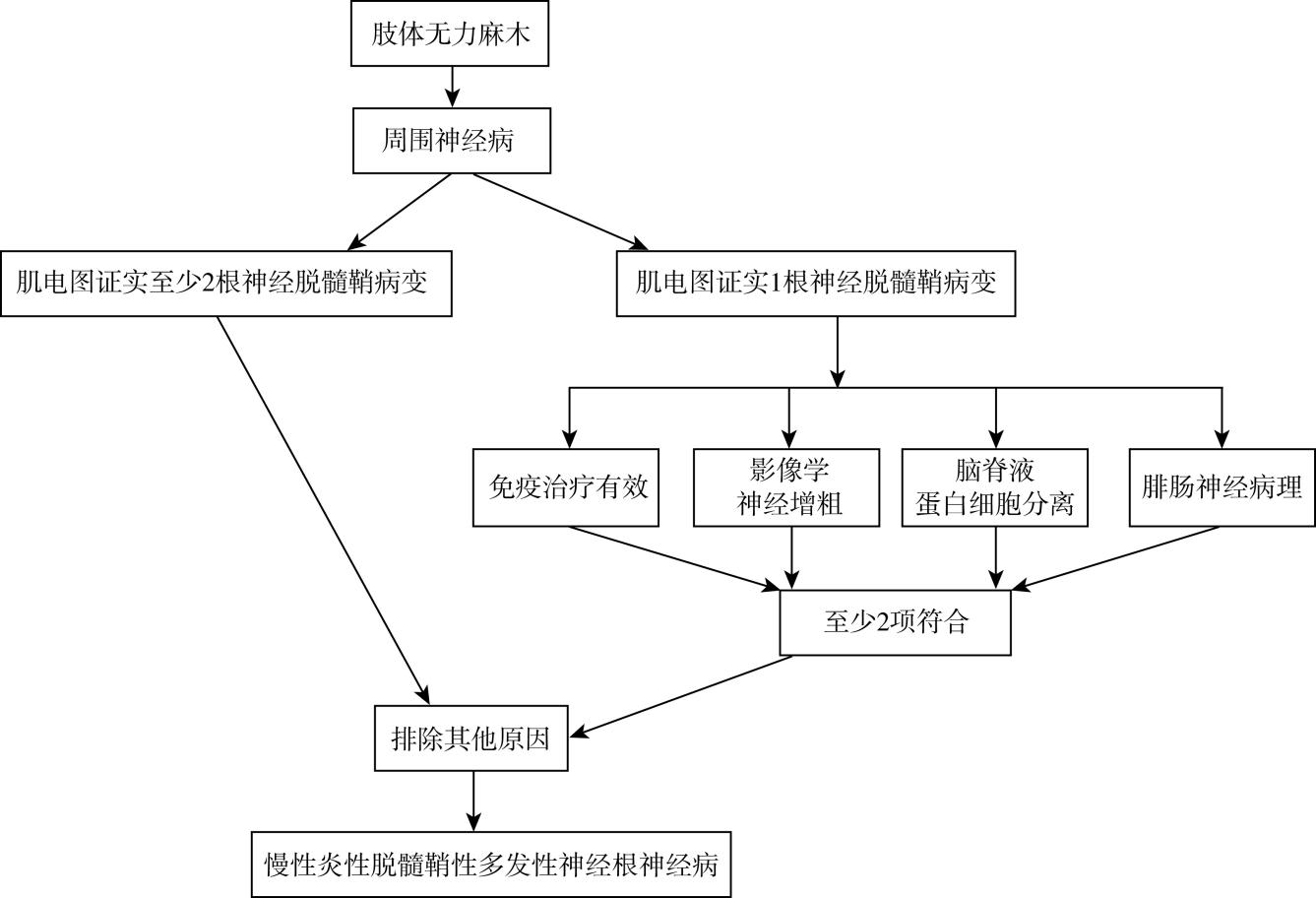
图13-1 慢性炎性脱髓鞘性多发性神经根神经病
参考文献
[1] VAN DEN BERGH PYK, VAN DOORN PA, HADDEN RDM, et al. European Academy of Neurology/Peripheral Nerve Society guideline on diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy: report of a joint task force-second revision. Eur J Neurol,2021,28(11):3556-3583.
[2]刘明生,崔丽英,蒲传强,中华医学会神经病学分会周围神经病协作组.慢性炎性脱髓鞘性多发性神经根神经病诊治中国专家共识2022.中华神经科杂志,2023,56(2): 125-132.
[3] BROERS MC, BUNSCHOTEN C, NIEBOER D, et al. Incidence and prevalence of chronic inflammatory demyelinating polyradiculoneuropathy: a systematic review and meta-analysis. Neuroepidemiology,2019,52(3-4):161-172.
[4] NIU J, LI Y, LIU T, et al. Serial nerve ultrasound and motor nerve conduction studies in chronic inflammatory demyelinating polyradiculoneuropathy. Muscle Nerve, 2019,60(3):254-262.
[5] DELMONT E, BRODOVITCH A, KOUTON L, et al. Antibodies against the node of Ranvier: a real-life evaluation of incidence, clinical features and response to treatment based on a prospective analysis of 1500 sera. J Neurol,2020,267(12):3664-3672.
[6] Allen JA, Lin J, Basta I, et la. Safety, tolerability, and efficacy of subcutaneous efgartigimod in patients with chronic inflammatory demyelinating polyradiculoneuropathy (ADHERE): a multicentre, randomised-withdrawal, double-blind, placebo-controlled, phase 2 trial. Lancet Neurol 2024; 23: 1013–1024