


指南与共识 | Ⅰ型干扰素病诊疗中国专家共识
中国罕见病联盟
Ⅰ型干扰素病是一类可累及全身多个脏器的自身炎症性疾病,具有较高致残率和致死率,严重影响患儿的生活质量,给家庭和社会带来负担。其临床表型谱宽泛,早期识别较为困难,诊断及治疗的延误会导致病情的加重且难以逆转,影响预后。中国儿童风湿免疫病联盟及中国药师协会罕见病用药工作委员会制定该共识,旨在对Ⅰ型干扰素病的定义、基因分类、临床表现、诊断及治疗给予相应的建议。
Ⅰ型干扰素病是一种单基因自身炎症性疾病,可累及皮肤、神经等全身多系统,自然病程下,具有较高的致残率和死亡率。随着近年来诊断技术的提高,越来越多的Ⅰ型干扰素病被发现。目前国内尚无关于Ⅰ型干扰素病的临床指南或专家共识,中国医师对于Ⅰ型干扰素病认识尚不足,加强对该疾病的早期临床识别能力和提高诊疗水平具有重要临床意义。因此中国儿童风湿免疫病联盟及中国药师协会罕见病用药工作委员会专家共同制定了该共识,旨在对Ⅰ型干扰素病的定义、基因分类、临床表现、诊断及治疗给予相应建议。
1 共识制定方法
本共识由中国儿童风湿免疫病联盟及中国药师协会罕见病用药工作委员会专家共同发起。共识专家组对共识推荐意见通过德尔菲法达成共识。共识专家组主要由儿童风湿免疫专家组成,共同讨论了共识的总体框架,拟定关键问题及共识提纲,初步确定覆盖了疾病的定义、临床表现、诊断及鉴别诊断、治疗、长期随访及管理的23个问题,经过讨论及投票,最终选定9个临床问题。以 “Type Ⅰ interferonopathy”“Ⅰ型干扰素病” 为主题词检索了Pubmed、Embase、Cochrane Library、Clinical Trials、中国知网和万方数据知识服务平台,检索时间为建库至2024年8月30日,筛选证据等级相对较高的文献,文献纳入标准为①研究对象为Ⅰ型干扰素病;②研究目的为分析Ⅰ型干扰素病的临床表现、辅助检查、诊断及治疗方案。除外主题不符及预警杂志的文章。对文献提取关键数据,并进行汇总分析,回答上述9个问题,形成初步共识建议。将初稿返回各位专家征求意见,根据反馈意见,修改共识草案,调整至8个建议。最后再次召集专家团队讨论,整改为7条推荐意见,经过17位专家投票形成共识度。投票等级分为:同意、 不同意,如“同意”票数超过60%,则达成共识,共识 度为“同意”所得票数占所有收回有效票数的百分比。
2 共识推荐意见
2.1 Ⅰ型干扰素病的定义
推荐意见:Ⅰ型干扰素病是一组由单基因缺陷导致Ⅰ型干扰素(interferon type Ⅰ,IFN-Ⅰ)信号通路增强的遗传性疾病,属于自身炎症性疾病,基于国际免疫学联合会 (International Union of Immunological Societies,IUIS)2022年免疫出生缺陷(inborn errors of immunity,IEI)分类共包含20种基因型(共识度100%)。
人类的IFN-Ⅰ包括IFN-α、-β、-ε、-κ、-ω,其中最常见的类型为IFN-α和IFN-β,具有较强的抗病毒活性。在模式识别受体识别外源性核酸后,受感染的细胞分泌IFN-Ⅰ,以自分泌或旁分泌模式作用于IFN-Ⅰ受体(IFN-α receptor,IFNAR),通过Janus激酶-信号转导和转录激活因子(Janus kinase-signal transducers and activators of the transcription,JAK-STAT)途径诱导干扰素刺激基因(interferon stimulat-ing genes,ISGs)的转录,增强细胞IFN信号通路,该过程存在相应的负反馈调节机制,以避免IFN信号通路过度活化导致细胞凋亡和坏死[1]。
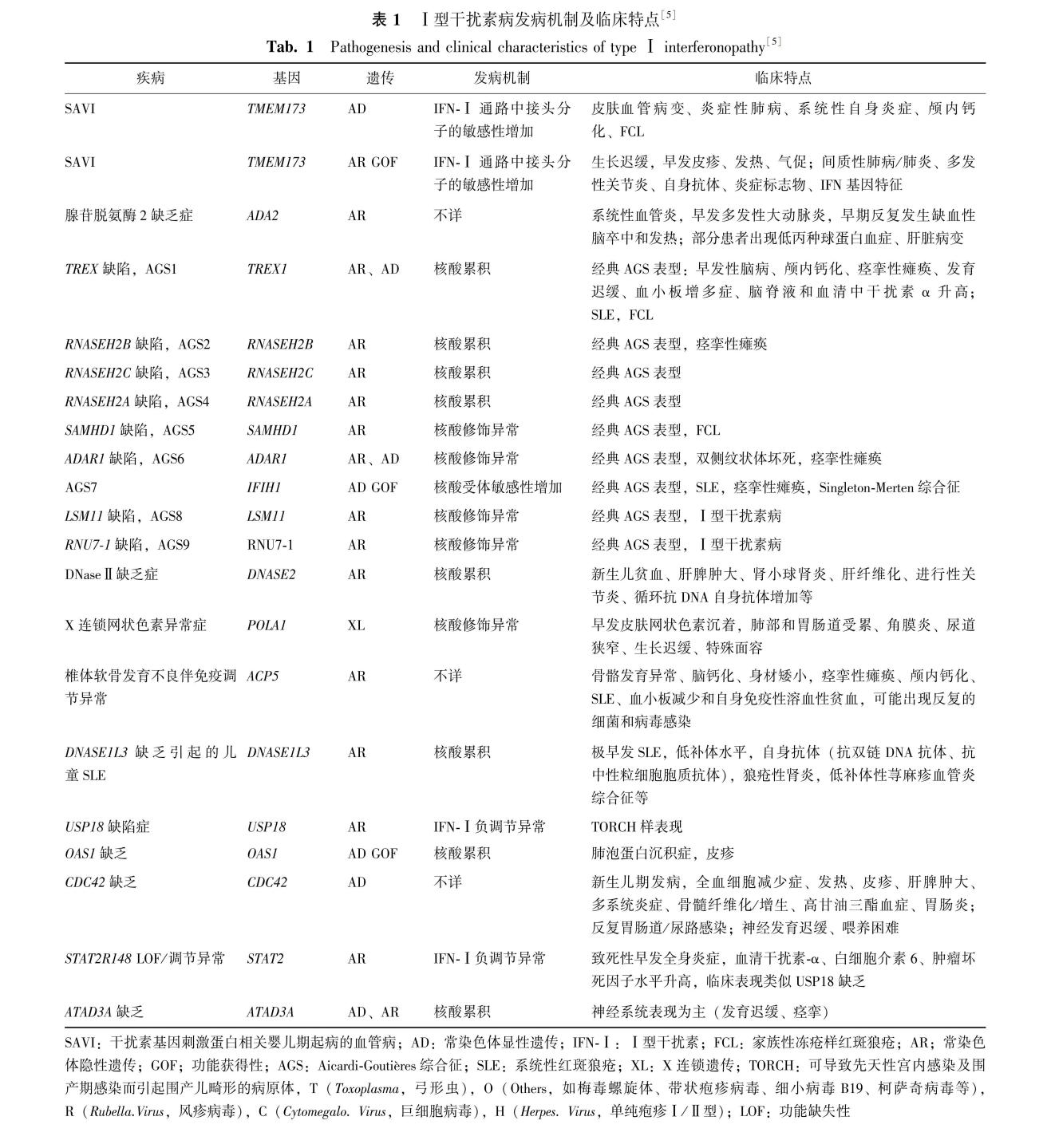
Ⅰ型干扰素病的概念是在2011年由Crow首次提出[2],是一组由IFN-Ⅰ信号通路过度活化引起的单基因遗传性疾病[3]。根据IUIS 2022年IEI分类,Ⅰ型干扰素病是IEI的一个亚组,详见表1。Ⅰ型干扰素病可能涉及的发病机制包括:核酸代谢异常、核酸受体敏感性增加或自发激活、IFN-Ⅰ 通路中接头分子的敏感性增加、IFN-Ⅰ 通路的负调节受干扰等。
基于IUIS 2022版IEI的分类,腺苷脱氨酶2缺乏症(deficiency of adenosine deaminase2,DADA2)目前仍被分到Ⅰ型干扰素病,然而其Ⅰ型IFN信号上调水平通常不如在其他Ⅰ型干扰素病中高,这种疾病被归类为Ⅰ型干扰素病可能不太合适。有研究认为蛋白酶体病归为Ⅰ型干扰素病[4],其相关基因包括PSMB8、PSMB4、PSMB9、PSMA3、POMP、PSMG2等,而IUIS将其归为非炎症小体病[5]。近些年还发现了一些可能与Ⅰ型干扰素病相关的新基因,包括REXO2[6]、ARF1[7]、RELA[8]、SAMD9L[9]、JAK1[10]、PNPT1[11]等,其发病机制尚需进一步研究。
2.2 Ⅰ型干扰素病的早期识别与诊断
推荐意见:Ⅰ型干扰素病可累及全身多个脏器,具有自身免疫性疾病和全身高炎症的特点,该类疾病发病年龄较早,结节性红斑、冻疮样皮疹、肌张力异常、基底节钙化、甲状腺功能异常、肺间质病变、血细胞减少等临床表现对于Ⅰ型干扰素病具有提示意义,其诊断则需要根据典型临床表现及基因检测结果进行诊断(共识度100%)。
Ⅰ型干扰素病可累及全身多个脏器,具有自身免疫性疾病和全身高炎症的特点[12]。神经系统最常见的症状包括肌张力异常、癫痫、脑血管事件等,头颅影像学可以表现为颅内钙化、脑白质病变、脑萎缩等。皮肤受累表现多为冻疮样皮疹、网状青斑、红斑、结节性红斑、雷诺现象、脂膜炎、肢端坏疽、溃疡、皮肤坏死等。其他临床表现还包括关节挛缩、肺间质病变、肺纤维化、肝脾肿大、肝酶升高、血细胞减少、自身抗体阳性、炎症指标升高、生长发育落后、甲状腺功能异常等[1,13]。
Ⅰ型干扰素病发病较早,80%以上在3岁前起病,由于其临床表型谱广泛,早期临床识别困难[14]。2019年Sonmez等[15]制定出识别Ⅰ型干扰素病的评分方法,共有8项,如符合其中3项可怀疑Ⅰ型干扰素病:①皮肤表现(结节性红斑、寒冷相关的肢体末端紫斑);②血管病变(冻疮样皮疹、微血管病变、肢端坏疽/溃疡/梗死);③脂肪营养不良;④关节表现(关节挛缩、非侵蚀性关节炎);⑤肌炎(局灶性);⑥神经系统异常(基底神经节钙化、脑白质营养不良、脑白质病变、脑脊液淋巴细胞反应);⑦肺部受累(间质性肺病、肺纤维化、肺动脉高压);⑧发作性白细胞或淋巴细胞减少。
尽管具有相似的发病机制,不同Ⅰ型干扰素病具有不同临床特点。如典型的Aicardi-Goutières综合征(Aicardi-Goutières syndrome,AGS)多在生后几天或几个月内起病,主要表现为早发性脑病、基底节钙化、白质异常、脑萎缩、痉挛性瘫痪、精神运动性退化或延迟、冻疮样皮疹、小头畸形、发育落后等[16]。干扰素基因刺激蛋白相关婴儿期起病的血管病(stimulator of interferon genes associated vasculopathy, infantile-onset, SAVI)的特征是早发性全身炎症伴反复发热、坏死性皮肤血管病变、多种皮疹和肺间质病变[17]。椎体软骨发育不良伴免疫调节异常(spondyloenchondrodysplasia with immune dysregulation,SPENCDI)的特征是以脊椎骨和干骺端发育异常、不同程度的神经功能障碍(如痉挛状态、发育迟缓和基底节钙化),此类患者易发生溶血性贫血、自身免疫性甲状腺炎、系统性红斑狼疮或自身免疫性肝炎等自身免疫性疾病[18]。DADA2的临床特征是系统性血管炎、血细胞减少、骨髓衰竭及免疫缺陷[19]。详见表1。
到目前为止,Ⅰ型干扰素病尚无临床诊断标准,主要根据典型临床表现及基因结果进行诊断。但有一部分患者,临床高度怀疑Ⅰ型干扰素病,基因检测为阴性,对于此类患者,应转诊至有经验的医疗机构进行诊疗。
2.3 Ⅰ型干扰素病的鉴别诊断
推荐意见:Ⅰ型干扰素病需要同先天性TORCH[指一组可导致先天性宫内感染及围产期感染而引起围产儿畸形的病原体:T(Toxoplasma,弓形虫)、O(Others,如梅毒螺旋体、带状疱疹病毒、细小病毒B19、柯萨奇病毒等)、R(Rubella.Virus,风疹病毒)、C(Cytomegalo.Virus,巨细胞病毒)、H(Herpes.Virus,单纯疱疹Ⅰ/Ⅱ型)]感染、其他自身炎症性疾病、风湿免疫性疾病等进行鉴别,同时不同Ⅰ型干扰素病之间需要相互鉴别(共识度100%)。
Ⅰ型干扰素病还需同其他疾病鉴别,如:①先天性TORCH感染,可以表现为生后血细胞减少、肝酶升高、颅内钙化等症状[20],可以结合母亲孕期感染病史及相关病毒学进行鉴别诊断;②其他自身炎症性疾病,如炎症小体病、非炎症小体病,均发病较早,累及全身多个系统,具有全身炎症反应[5]。但Ⅰ型干扰素病中IFN水平明显升高,而另两类疾病中通常无明显升高,但仍需基因检测进行明确诊断;③自身免疫性疾病,如系统性红斑狼疮、混合结缔组织病、原发性干燥综合征等[21],起病相对较晚,多在青春期起病,具有多种特异性自身免疫抗体,且应用常规的激素、免疫抑制剂治疗有效。另外,不同的Ⅰ型干扰素病之间也需要相互鉴别诊断。
2.4 Ⅰ型干扰素病的主要辅助检查的选择
推荐意见:因Ⅰ型干扰素病可累及全身多个脏器,对于初次诊断的患儿,应进行全面评估。建议至有条件的医疗机构应用定量聚合酶链反应(quantitative polymerase chain reaction,qPCR)检测ISGs的mRNA表达水平,从而协助诊断Ⅰ型干扰素病。对于怀疑Ⅰ型干扰素病的患者均应行基因检测,首先推荐全外显子或全基因组测序,还可应用覆盖目前已知的IEI致病基因的基因panel进行测序,Sanger测序可用于高度怀疑某种Ⅰ型干扰素病的情况(共识度100%)。
2.4.1 一般辅助检查
体格发育情况包括身高、体重、头围;常规实验室检查包括红细胞沉降率、C反应蛋白等炎症指标,以及血常规、肝肾功、甲状腺功能、尿常规、免疫球蛋白、淋巴细胞亚群、自身抗体、血脂、肌酶等化验。进一步的辅助检查根据临床需求可完善肝胆胰脾肾超声、头颅CT、头颅MRI、脑电图、肺部高分辨CT、肺功能、超声心动图、心电图、智力发育评估,如怀疑血管受累,还需完成相应血管的超声或造影检查;耳鼻喉科完善听力评估,眼科完善视力、眼底、眼压评估。如有需要,还可进一步完善骨髓穿刺、肾脏穿刺、腰椎穿刺等有创检查[22]。
2.4.2 干扰素信号检测
2.4.2.1 单分子阵列检测应用 单分子阵列检测(single-molecule array,SIMOA)可以直接检测IFN-α蛋白的表达水平[23],但由于在不同的实验中可重复性差,且设备尚未普及,SIMOA 在常规使用中受到限制。
2.4.2.2 qPCR 应用qPCR[24-25]检测ISGs的mRNA表达水平,国外也有应用Nanostring技术测定ISGs的报道[26],但国内尚未开展。这些检查不具有疾病特异性,除Ⅰ型干扰素病外,一些自身免疫性疾病,如系统性红斑狼疮和皮肌炎也会表现ISGs表达升高[27]。同时ISGs表达水平与临床状态之间的相关性较差,例如一部分无临床症状的个体表现出干扰素信号明显上调,而在部分Ⅰ型干扰素病患者中,ISGs水平具有年龄相关性,在4~5岁之后可自行下降[13]。因此不能单独依靠ISGs表达水平来诊断或除外Ⅰ型干扰素病。
2.4.2.3 二代测序 对于临床怀疑Ⅰ型干扰素病的患者,推荐使用二代测序来筛查致病基因[28]。近年来,由于一些新的Ⅰ型干扰素病致病基因被不断发现,建议首先选择全外显子组测序,必要时行全基因组测序。一些自身炎症性疾病基因panel,可用于诊断目前已发现基因所致的疾病,对于探索新的基因有一定的限制。如临床诊断高度倾向某种Ⅰ型干扰素病,也可行Sanger测序[21]。
目前大多数Ⅰ型干扰素病相关基因突变为点突变或小插入-缺失变异,少部分基因也可出现嵌合体,如STING1基因[29],对于这些疾病,需应用尿液、口腔鼻黏膜细胞、毛发等多种组织进行测序验证。迄今为止,Ⅰ型干扰素病中尚无拷贝数变异的报道。在基因诊断的过程中,需考虑上述特殊情况。
2.5 Ⅰ型干扰素病的治疗建议
推荐意见:治疗原则是早期干预,控制症状,减少并发症,减少与治疗相关的副作用或毒性。JAK抑制剂(JAK inhibitor,JAKi)可以阻断IFN-Ⅰ信号通路,临床上已用于治疗Ⅰ型干扰素病。造血干细胞移植可用于部分Ⅰ型干扰素病的治疗,如DADA2(共识度100%)。
2.5.1 治疗原则
尽量早期干预,控制临床症状,阻止或延缓疾病进展,减少脏器损伤,尽量减少与治疗相关的不良反应[22]。Ⅰ型干扰素病应用常规激素及免疫抑制剂治疗效果不佳。根据其发病机制,目前有以下几种潜在的治疗方法。
2.5.2 JAK抑制剂
JAKi可通过抑制JAK阻断IFN-Ⅰ信号通路[30]。此类药物已被报道用于治疗SAVI、SPENCDI和AGS等疾病[30-34],不同研究选择的剂量及临床治疗效果均不相同,且多数为病例报道。Vanderver等[35]于2020 年进行了一项单中心、开放标签试验,共纳入35例接受巴瑞替尼[0.1~0.6 mg/(kg·d)]治疗的AGS患者,结果显示治疗对全身,尤其是皮肤症状有显著效果,部分患儿的发育里程碑也得到改善。Frémond等[32]也进行了巴瑞替尼[0.5~0.8 mg/(kg·d)]及芦可替尼[12~30 mg/(m2·d)]治疗AGS的单中心真实世界研究,结果相似,但是神经系统症状改善不明显。一篇关于芦可替尼[0.25~0.71 mg/(kg·d)]及托法替布[0.2~0.5 mg/(kg·d)]治疗Ⅰ型干扰素病的研究,共纳入2例SAVI 患者、1例AGS1患者、1例AGS7患者和2例SPENCDI患者,JAKi可改善皮肤病变,减少发热,降低ISGs水平[36]。因此,使用 JAKi可降低ISGs的水平,改善皮肤炎症,但由于只有少量药物可透过血脑屏障,所以此类药物对神经系统症状的改善并不明显[32]。
2.5.3 IFNAR阻滞剂
阿尼鲁单抗可结合IFNAR1,阻止受体和配体复合物的形成,阻断JAK-STAT激活和ISGs的转录。在国外已有阿尼鲁单抗治疗SAVI、CANDLE综合征、DNase Ⅱ缺乏症患者的报道[37-38],该药可有效缓解临床症状,降低ISGs转录水平,抑制炎症反应,改善生长发育。
2.5.4 逆转录酶抑制剂
内源性核酸在AGS发病机制中起重要作用,核酸的积累可能来自内在核酸代谢或内源性逆转录因子障碍。根据此假设,使用逆转录酶抑制剂(reverse transcriptase inhibitor,RTI)对于AGS患者可能有益。2018年,一项纳入8例AGS患者的单中心、开放标签的临床试验,联合应用三种RTI(阿巴卡韦、拉米夫定和齐多夫定)治疗,研究时间为12个月,该研究表明RTI可有效降低患儿血液、脑脊液中的Ⅰ型干扰素信号强度[39]。其具体治疗效果尚需进一步探索。
2.5.5 造血干细胞移植
目前造血干细胞移植(hematopoietic cell transplantation,HSCT)用于治疗Ⅰ型干扰素病的报道较少,在Signa等[40]发表的一篇评论中总结分析了100例患者接受了HSCT的治疗效果,其中Ⅰ型干扰素病患者47例,包括蛋白酶体病患者5例、SAMD9L基因突变患者4例、STAT2基因突变患者1例、OAS1缺乏症患者4例、DADA2患者33例,接受HSCT治疗后,结果各不相同,完全缓解38例,部分缓解3例,缓解多以过度炎症及自身免疫反应为主,但神经系统症状仍在进展。6例死亡,多死于严重并发症。因此HSCT可能纠正与Ⅰ型干扰素病相关的炎症反应及免疫功能障碍,但对神经系统症状的改善仍有待商榷。整体来说,应用HSCT治疗Ⅰ型干扰素病的经验相对较少,仍需大样本研究。
2.5.6 其他药物治疗
TNF-α抑制剂常规用于DADA2的治疗,对于抑制炎症反应及血管病变有显著效果。2024年一项国外研究,共纳入32例DADA2患者,其中27例应用TNF-α抑制剂治疗,随访中位时间为58个月,10例以皮肤及神经系统受累的患者(35.7%)完全缓解,10例患者 (35.7%)部分缓解,未接受抗TNF治疗的7例患者中,死亡5例,拒绝治疗1例,1例在骨髓移植后治愈[41]。此外也有IL-6抑制剂治疗AGS7[42]、AGS5[43]的病例报道,结果提示IL-6抑制剂可有效抑制全身炎症反应,以及逆转AGS5相关的脑血管病变。国内还有沙利度胺治疗AGS、DADA2的报道[44],沙利度胺可降低炎症指标及疾病活动度。但均缺乏大数据研究。
2.6 Ⅰ型干扰素病的疾病严重程度及治疗效果评估方法
推荐意见:目前缺乏统一的病情评估方法,多以对临床症状及辅助检查的描述为主,以及炎症指标、ISGs评分的高低来评估病情及治疗效果(共识度95.65%)。
因Ⅰ型干扰素疾病谱较庞大,且不同疾病之间临床差异性较大,目前尚无统一的量化标准进行疾病严重程度的评估,只有少数研究有特异的评分,如AGS评分、AGS每日评分、CANDLE每日评分、SAVI每日评分等[30,35,45]。其他Ⅰ型干扰素病评估病情多以对临床症状及辅助检查的描述为主,以及炎症指标、ISG评分的高低来评估病情及治疗效果。
2.7 Ⅰ型干扰素病患者的长期管理
推荐意见:长期随访过程中需要关注各系统受累情况及患儿的生长发育情况,每次就诊时均应评估所有患者是否存在疾病特异性症状、全身炎症反应及药物相关副作用,根据结果调整治疗。建议患儿的家系成员在生育前行遗传咨询(共识度100%)。
应根据疾病活动度和严重程度确定随访频率,关注各系统受累情况及患儿的生长发育情况,根据病情变化及时调整治疗方案,密切监测疾病进展,每次就诊时均应评估所有患者是否存在疾病特异性症状和全身炎症。同时还要监测JAKi的相关副作用,JAKi相关的不良事件包括血细胞减少、泌尿道和上呼吸道感染、疱疹病毒再激活、肝功能异常和高胆固醇血症。更严重和罕见的不良事件包括血栓栓塞事件、乙型肝炎病毒再激活、播散性结核病、胃肠道穿孔和恶性肿瘤[22]。
最后建议患儿的家系成员再生育做遗传咨询,需结合遗传模式、患儿变异基因的来源、临床是否外显等多种因素综合考虑。
3 结语
本共识结合国外最新研究进展及国内相关领域专家的临床实践经验,为中国Ⅰ型干扰素病的临床诊疗提供了重要参考。但由于此类疾病发病率相对较低,缺乏大队列随机对照试验研究,多为病例报道或病例系列分析,证据等级较低,且国内文献相对较少,因此本共识具有一定局限性,尚需更多的基础实验及临床研究来完善。
作者贡献:本专家共识由中国儿童风湿免疫病联盟、中国药师协会罕见病用药工作委员会发起,宋红梅牵头成立共识制订工作组,编写组成员共同起草了共识初稿,专家组成员共同进行讨论和投票。
利益冲突:所有参与本共识制订的人员均声明不存在利益冲突。
执笔专家:马明圣(北京协和医院),王伟(北京协和医院)
核心专家组成员(以姓氏拼音为序):杜悦(中国医科大学附属盛京医院),李小青(西安交通大学附属儿童医院),卢美萍(浙江大学医学院附属儿童医院),戎赞华(河北医科大学第二医院),宋红梅(北京协和医院),孙利(复旦大学附属儿科医院),唐雪梅(重庆医科大学附属儿童医院),吴小川(中南大学湘雅二医院),杨军(深圳市儿童医院),杨思睿(吉林大学第一医院),俞海国(南京市儿童医院),曾萍(广州市妇女儿童医疗中心),赵冬梅(乌鲁木齐儿童医院),张建江(郑州大学第一附属医院),张秋业(青岛大学附属医院),张伟(成都市妇女儿童中心医院)
秘书成员:张天誉(北京协和医院),周煜(北京协和医院)
致谢:张丁丁(北京协和医院临床研究所)
参考文献
[1]Crow YJ, Stetson DB. The type Ⅰ interferonopathies: 10 years on[J]. Nat Rev Immunol,2022,22(8):471-483.
[2]Crow YJ. Type Ⅰ interferonopathies: a novel set of inborn errors of immunity[J]. Ann N Y Acad Sci,2011,1238:91-98.
[3]Crow YJ, Casanova JL. Human life within a narrow range: the lethal ups and downs of type Ⅰ interferons[J]. Sci Immunol,2024,9(97):eadm8185.
[4]Cetin Gedik K, Ortega-Villa AM, Materne G, et al. Disease flares with baricitinib dose reductions and development of flare criteria in patients with CANDLE/PRAAS[J]. Ann Rheum Dis,2024,83(9):1181-1188.
[5]Tangye SG, Al-Herz W, Bousfiha A,et al. Human inborn errors of immunity: 2022 update on the classification from the International Union of Immunological Societies Expert Committee[J]. J Clin Immunol,2022,42(7):1473-1507.
[6]Idiiatullina E, Al-Azab M, Lin M, et al. Heterozygous de novo dominant negative mutation of REXO2 results in interferonopathy[J]. Nat Commun,2024,15(1):6685.
[7]Hirschenberger M, Lepelley A, Rupp U, et al. ARF1 prevents aberrant type Ⅰ interferon induction by regulating STING activation and recycling[J]. Nat Commun,2023,14(1):6770.
[8]Moriya K, Nakano T, Honda Y, et al. Human RELA dominant-negative mutations underlie type Ⅰ interferonopathy with autoinflammation and autoimmunity[J]. J Exp Med,2023,220(9):e20212276.
[9]Kanazawa N, Ishii T, Takita Y, et al.Efficacy and safety of baricitinib in Japanese patients with autoinflammatory type Ⅰ interferonopathies (NNS/CANDLE, SAVI, And AGS)[J]. Pediatr Rheumatol Online J,2023,21(1):38.
[10]Rodero MP, Crow YJ. Type Ⅰ interferon-mediated mono-genic autoinflammation: the type Ⅰ interferonopathies, a conceptual overview[J]. J Exp Med,2016,213(12):2527-2538.
[11]Vedrenne V, Gowher A, De Lonlay P, et al. Mutation in PNPT1, which encodes a polyribonucleotide nucleotidyltransferase, impairs RNA import into mitochondria and causes respiratory-chain deficiency[J]. Am J Hum Genet,2012,91(5):912-918.
[12]Haslak F, Kilic H, Sahin S,et al. Children with type 1 interferonopathy: commonalities and diversities in a large patient cohort[J]. J Rheumatol,2024.doi: 10.3899/jrheum.2024-0294.
[13]Rice G, Patrick T, Parmar R, et al. Clinical and molecular phenotype of Aicardi-Goutieres syndrome[J]. Am J Hum Genet,2007,81(4):713-725.
[14]王伟,王薇,邹丽萍,等.Ⅰ型干扰素病25例临床特点分析[J].中华儿科杂志,2021,59(12):1043-1047.
[15]Sonmez HE, Karaaslan C, de Jesus AA, et al.A clinical score to guide in decision making for monogenic type Ⅰ IFNopathies[J]. Pediatr Res,2020,87(4):745-752.
[16]Crow YJ. Aicardi-Goutières Syndrome[EB/OL].(2005-06-29)[2024-08-22].https://www.ncbi.nlm.nih.gov/books/NBK1475/.
[17]Frémond ML, Hadchouel A, Berteloot L, et al. Overview of STING-associated vasculopathy with onset in infancy (SAVI) among 21 patients[J]. J Allergy Clin Immunol Pract,2021,9(2):803-818.e11.
[18]Kara B, Ekinci Z, Sahin S, et al.Monogenic lupus due to spondyloenchondrodysplastia with spastic paraparesis and intracranial calcification: case-based review[J]. Rheumatol Int,2020,40(11):1903-1910.
[19]Wouters M, Ehlers L, Dzhus M, et al. Human ADA2 deficiency: ten years later[J]. Curr Allergy Asthma Rep,2024,24(9):477-484.
[20]Meuwissen ME, Schot R, Buta S, et al. Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome[J]. J Exp Med,2016,213(7):1163-1174.
[21]Wang W, Yu Z, Gou L, et al. Single-center overview of pediatric monogenic autoinflammatory diseases in the past decade: a summary and beyond[J]. Front Immunol,2020,11:565099.
[22]Cetin Gedik K, Lamot L, Romano M, et al. The 2021 European Alliance of Associations for Rheumatology/American College of Rheumatology Points to Consider for Diagnosis and Management of Autoinflammatory Type Ⅰ Interferonopathies: CANDLE/PRAAS, SAVI, and AGS[J]. Arthritis Rheumatol,2022,74(5):735-751.
[23]Rodero MP, Decalf J, Bondet V, et al. Detection of interferon alpha protein reveals differential levels and cellular sources in disease[J]. J Exp Med,2017,214 (5): 1547-1555.
[24]Rice GI, Forte GM, Szynkiewicz M, et al. Assessment of interferon-related biomarkers in Aicardi-Goutières syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: a case-control study[J]. Lancet Neurol,2013,12(12):1159-1169.
[25]李文道, 王薇, 王伟, 等. 儿童干扰素刺激基因表达检测方法的建立与应用[J] .中华检验医学杂志,2022,45(6) : 603-609.
[26]de Jesus AA, Hou Y, Brooks S, et al. Distinct interferon signatures and cytokine patterns define additional systemic autoinflammatory diseases[J]. J Clin Invest,2020,130(4):1669-1682.
[27]Rodríguez-Carrio J, Burska A, Conaghan PG, et al. 2022 EULAR points to consider for the measurement, reporting and application of IFN-Ⅰ pathway activation assays in clinical research and practice[J]. Ann Rheum Dis,2023,82(6):754-762.
[28]Peng X, Kaviany S. Approach to diagnosing inborn errors of immunity[J]. Rheum Dis Clin North Am,2023,49(4):731-739.
[29]de Becdelièvre A, Eveillard LA, Wolska-Kus'nierz B,et al. Inheritance of STING mosaicism in two half-siblings[J]. J Clin Immunol,2024,44(7):168.
[30]Sanchez GAM, Reinhardt A, Ramsey S, et al. JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies[J]. J Clin Invest,2018,128(7):3041-3052.
[31]Boyadzhieva Z, Ruffer N, Burmester G, et al.Effectiveness and safety of JAK inhibitors in autoinflammatory diseases: a systematic review[J]. Front Med (Lausanne),2022,9:930071.
[32]Frémond ML, Hully M, Fournier B, et al. JAK inhibition in Aicardi-Goutières syndrome: a monocentric multidisciplinary real-world approach study[J]. J Clin Immunol,2023,43(6):1436-1447.
[33]Wu J, Zhou Q, Zhou H, et al. Case report: JAK1/2 inhibition with baricitinib in the treatment of STING-associated vasculopathy with onset in infancy[J]. Pediatr Rheumatol Online J,2023,21(1):131.
[34]Gernez Y, Narula M, Cepika AM, et al. Case report: refractory evans syndrome in two patients with spondyloenchondrodysplasia with immune dysregulation treated successfully with JAK1/JAK2 inhibition[J]. Front Immunol,2024,14:1328005.
[35]Vanderver A, Adang L, Gavazzi F, et al. Janus kinase inhibition in the Aicardi-Goutières syndrome[J]. N Engl J Med,2020,383(10):986-989.
[36]Li W, Wang W, Wang W, et al. Janus kinase inhibitors in the treatment of type Ⅰ interferonopathies: a case series from a single center in China[J]. Front Immunol,2022,13:825367.
[37]Kretzschmar G, Páez LP, Tan Z, et al. Normalized interferon signatures and clinical improvements by IFNAR1 blocking antibody (Anifrolumab) in patients with type Ⅰ interferonopathies[J]. J Clin Immunol,2024,45(1):31.
[38]Doroudchi MA, Thauland TJ, Patel BA, et al. Anifrolumab to treat a monogenic interferonopathy[J]. J Allergy Clin Immunol Pract,2024,12(5):1374-1376.
[39]Rice GI, Meyzer C, Bouazza N, et al. Reverse-transcriptase inhibitors in the Aicardi-Goutières syndrome[J].N Engl J Med, 379(23):2275-2277.
[40]Signa S, Dell'Orso G, Gattorno M, et al. Hematopoietic stem cell transplantation in systemic autoinflammatory diseases-the first one hundred transplanted patients[J].Expet Rev Clin Immunol,2022,18 (7) : 667-689.
[41]Ayan G, Basaran O, Firlatan B, et al. Long-term follow-up of anti-TNF treatment in adult and pediatric DADA2 patients: insights from real-world data[J].Int J Rheum Dis,2024,27(11):e15377.
[42]Wang W, Wang W, Peng S, et al. Tocilizumab reduces the unmanageable inflammatory reaction of a patient with Aicardi-Goutières syndrome type 7 during treatment with ruxolitinib[J].Pediatr Rheumatol Online J,2023,21(1):117.
[43]Henrickson M, Wang H. Tocilizumab reverses cerebral vasculopathy in a patient with homozygous SAMHD1 mutation[J].Clin Rheumatol,2017,36(6):1445-1451.
[44]Zhang C, Yu Z, Gao S, et al. Efficacy and safety of thalidomide in children with monogenic autoinflammatory diseases: a single-center, real-world-evidence study[J].Pediatr Rheumatol Online J,2023,21(1):124.
[45]Adang LA, Gavazzi F, Jawad AF, et al. Development of a neurologic severity scale for Aicardi Goutières Syndrome[J]. Mol Genet Metab,2020,130(2):153-160.