


146、面肩肱型肌营养不良症
罕见病诊疗指南(2025年版)
概述 面肩肱型肌营养不良症(facioscapulohumeral muscular dystrophy, FSHD;OMIM 158900)是继抗肌萎缩蛋白病(Duchenne型和Becker型肌营养不良)和强直性肌营养不良症(myotonic dystrophy,DM)之后最常见的肌营养不良症,为常染色体显性遗传。最早于1884年由法国医生Landouzy和Dejerine描述,又称Landouzy-Dejerine型肌营养不良症。1950年,Tyler和Stephens对一个犹太人大家系的临床特点进行详细调查,根据其特征性面部、肩胛带和上臂肌肉受累而正式命名为“Facioscapulohumeral Muscular Dystrophy”。主要表现为面肌、肩胛带肌和上肢肌群起病的无力和萎缩,逐渐累及躯干肌、骨盆带肌和下肢肌群,出现仰卧起坐不能、行走困难、脊柱畸形等,病程缓慢,预后相对较好,一般不影响寿命。2014年欧美报道其发病率约为(5~12)/10万,国内一项全国性调查的基因确诊患病率约为0.075/10万。
病因和流行病学
FSHD的基因结构和分子病理机制相对复杂,根据发病机制的差异分为 1型和2型,两者临床表型高度一致。FSHD1约占95%以上,不同于致病基因突变导致编码蛋白功能异常的经典模式,基因定位于4q35亚端粒区域一条多态性EcoRⅠ片段,由其内部长约3.3kb的D4Z4大卫星串联重复单位(D4Z4 repeats, DRs)多拷贝数缺失导致,属于特殊的基因结构变异(structural variation,SV)。正常人群EcoRⅠ片段长度为38~300kb,相应D4Z4拷贝数为11~100个,而患者的一条D4Z4拷贝数减少至1~10个,相应EcoRⅠ片段缩短至38kb以下。2型占比少于5%,在国内罕见报道,存在正常长度的4q35-DRs,主要由SMCHD1、DNMT3B和LRIF1基因突变导致。两型FSHD均与4q35-DRs上游3-kb处特异性单序列长度多态性片段(simple sequence length polymorphism,SSLP)和下游10kb处特殊的等位序列4qA/4qB存在密切连锁,只有特异性SSLP-4qA基因型才具有PAS(polyadenylation signals,PAS)尾端,以稳定末个D4Z4单元内DUX4基因表达而致病,中国患者主要为4A161PAS单体型,这些分子遗传学特征是进行基因检测的基础。4q35-DRs拷贝数减少或SMCHD1、DNMT3B和LRIF1基因存在致病性突变导致局部染色质结构失去稳定性,引起D4Z4广泛低甲基化改变及其DUX4基因获得性异常表达,产生氧化应激、炎症反应、缺氧诱导损伤等一系列级联病理生理反应,造成肌纤维凋亡和坏死,所以FSHD是一种表观遗传学效应导致的肌病,具体发病机制仍有待明确(图25-1)。
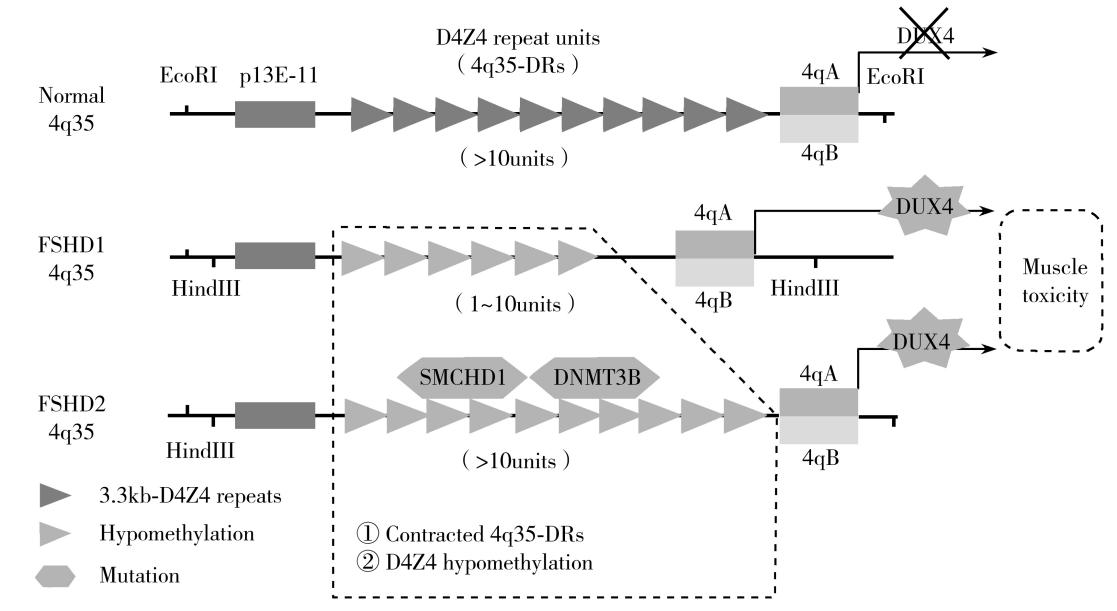
图25-1 FSHD分子遗传学机制模式图
在正常人中,一条染色体4q35的亚端粒区域包含大于10个高度紧密的(3.3kb)D4Z4大卫星重复序列;在FSHD患者中,该区域的D4Z4拷贝数缩短至1~10个,染色质结构松散和甲基化水平降低,引起末尾D4Z4序列内部的DUX4基因重新获得性表达。其中,1型D4Z4重复拷贝数目减少至1~10个,2型与SMCHD1、DNMT3B等基因突变有关,二者在结构上都需具有下游4qA等位序列和PAS尾端,稳定表达DUX4。E:EcoRⅠ 酶切位点;H:HindⅢ 酶切位点。
临床表现
FSHD的发病年龄、进展和严重程度存在家系间/家系内个体差异。经典型约占90%,通常在10~20岁或之后出现核心症状,另外约10%为早发型/严重型,在5岁之前出现面肌无力,10岁之前出现肩胛带肌无力和萎缩,多数为当代突变。有别于多数肌病,FSHD受累部位和程度多呈不对称性及“从上而下”缓慢性发展,从面肌、肩胛带肌和上臂肌群,依次发展到躯干肌、腰腹肌、骨盆带肌和下肢肌群。受累的面部肌肉包括眼轮匝肌和口轮匝肌,表现闭目、吹哨、鼓腮、噘嘴等动作困难,难以形成正常面部表情,笑容不自然而出现“肌病面容”,通常不影响日常生活,未被关注(图25-2A);肩胛带肌及上臂肌群受累表现为双臂抬举受限,不能将物体举过肩膀,出现“翼状肩胛”“蹼颈征”及“山峰征”(图25-2B、C),上肢远端受累表现为伸腕和伸指无力,肩-肱型肌无力和萎缩是常见的首诊症状;腰腹肌无力表现为仰卧起坐不能或下床困难,典型的“Beevor’s”征;容易受累的躯干肌还包括竖脊肌、胸大肌和腹直肌以及其他背肌,引起漏斗胸、腰椎前凸和脊柱侧弯等;受累的下肢肌肉包括大收肌、腘绳肌、臀肌、股四头肌和胫前肌,引起登梯、蹲立和足背屈无力,肌病步态及垂足征等表现。据国外数据表明,约23.7%的患者将发展至重度残疾——轮椅依赖状态,而国内相应的队列数据表明,12.0%的基因确诊的患者可能在起病后40年内丧失独立行走能力,依赖轮椅的发生率约为8.9%。除此还有不典型患者,例如面肌不受累患者(SHD)的表型相对较轻,以及相对少见的骨骼肌系统外症状,包括听力下降、视网膜血管病变、呼吸及循环系统病变、癫痫、智力低下等。
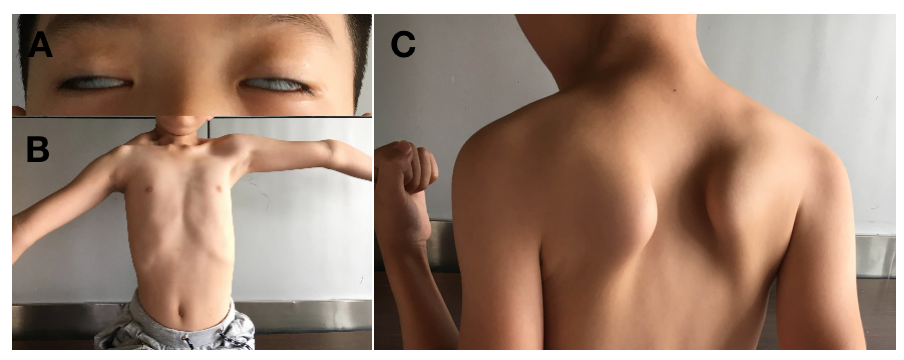
图25-2 FSHD典型体征
A:面肌无力;B:上肢抬举受限、蹼颈及山峰征;C:翼状肩胛。
FSHD临床表现和进展情况可以通过量表来评估,包括临床严重程度评分(clinical severity scoring,CSS)、临床评分(clinical scoring,CS)和年龄校正临床严重程度评分(ACSS=CSS × 2/就诊年龄× 1000),以及综合临床评估表(CCEF)被用识别典型/非典型患者。其临床异质性与多种因素有关,包括性别、D4Z4重复数及表观遗传差异等,女性丧失独立行走的发生率可能比男性更高,D4Z4重复数越小会导致更严重的表型。表观遗传效应也起重要作用,4q35-DRs低甲基化水平与疾病严重程度和残疾进展状态相关。此外,FSHD存在遗传早现现象,即家系成员代际之间4q35-DRs甲基化水平递降,肌无力严重程度递增,起病年龄提前,嵌合体型或代际间表观遗传改变可以部分解释。
辅助检查
1.实验室检查 多数患者肌酸激酶(CK)水平呈轻度至中度升高,很少超过1000 U/L,无诊断特异性,通常结合血清肌红蛋白(MYO)提示存在肌肉的受累情况。
2.肌电图 多数表现为肌源性损害,MNCS表现为CMAP幅低,SNCS无异常。
3.肌肉病理活检 早期通常出现散在的坏死、萎缩肌纤维和少数核内移,间质增生等轻度肌营养不良改变,缺乏特异性。在疾病的进展期,受累肌肉常伴有血管周围部位的炎症细胞浸润,晚期被大量间质脂肪和纤维组织替代。
4.肌肉影像 骨骼肌核磁共振(MRI)无创、低风险,可直观反应受累肌群的分布模式,显示常规体检无法检查到的肌肉,如脊旁肌或长内收肌,获得受累肌群的完整图像,协助鉴别诊断;判断肌肉的受累形态(萎缩或肥大)和程度,动态随访病情,监测疾病进展;T2-STIR+可以鉴别肌肉的炎症水肿、坏死或脂肪变,作为疾病进展的早期标志 。定量肌超声(QMUS)可以测量肌肉中的炎症、脂肪浸润和纤维化组织。利用超声检测患者的膈肌受累情况,结合肺功能检测能客观评估呼吸功能,可以作为临床常规检查。
5.基因检测 FSHD表型具有较高的临床辨识度,但基因检测有助于确诊,以经典的分子杂交法为基础,通过低熔点胶抽提基因组DNA,EcoRⅠ、HindⅢ、BlnⅠ酶切后脉冲电场凝胶电泳(pulsed field gel electrophoresis,PFGE)/Southern blotting印迹,标记p13E-11、4qA/4qB分子探针杂交,行限制性片段长度多态性(restriction fragment length polymorphism,RFLP)分析,结合靶向CpG甲基化检测,获得4q35-D4Z4重复数(即4q35-EcoRⅠ片段长度)及甲基化程度的分子遗传学诊断信息,这个检测体系是“金标准”,但相对复杂繁琐,周期长,难度大(图25-3)。近年来发展了Bionano光学图谱技术(optical genome mapping,OGM)等可提高检测效率,三代单分子检测技术(如纳米孔测序)也在探索中。考虑2型则需要结合二代测序进行SMCHD1、DNMT3B、LRIF1基因突变检测。
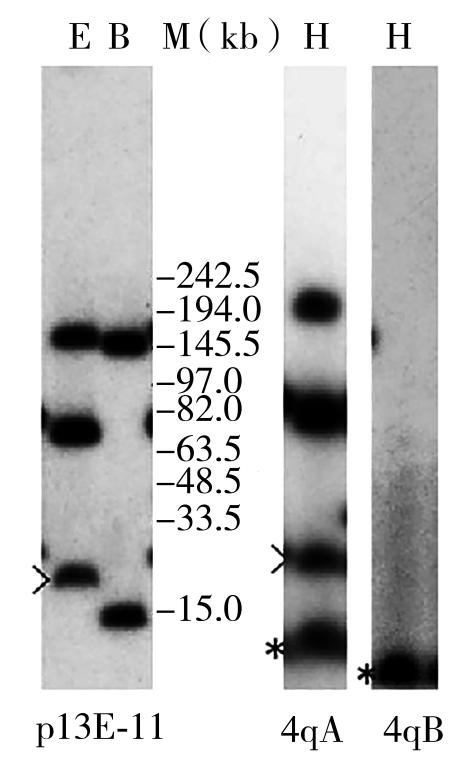
图25-3 FSHD分子杂交自显影检测图
基因组DNA酶切/PFGE电泳后,Southern blotting印迹和探针杂交显示存在缩短至17-kb的4qA型4q35 EcoRⅠ/p13E-11致病性片段。(E:EcoRⅠ酶切;B:EcoRⅠ/BlnⅠ双酶切;H:HindⅢ酶切;p13E-11:p13E-11分子探针杂交;4qA:4qA分子探针杂交;4qB:4qB分子探针杂交;M:MidRange PFG Marker I分子量标记;>:致病片段;*:非特异性片段。)
诊断
FSHD的临床辨识度较高,诊断需要结合特征性核心表型与家族史、实验室检查以及基因检测。诊断要点包括:
1.青少年或青年期起病为主,少数为儿童期或婴幼儿期,慢性病程。
2.不对称性肌病,表现为双上肢抬举困难或下肢无力,多数由上而下发展,伴肩胛骨突出;查体可见肌病面容,闭眼不全、鼓腮困难、鱼嘴征,翼状肩胛、蹼颈征、山峰征,Beevor’s征、肌病步态等典型体征。
3.部分有常显的家族史,早发型多为当代突变。
4.肌酶轻度升高,肌电图为肌源性损害,MRI为脂肪浸润、肌肉萎缩和炎性改变,肌肉病理为不同程度的肌营养不良改变,进展期伴血管周围炎性细胞浸润。
5.基因检测 FSHD1患者4q35-EcoRⅠ片段缩短至38 kb以下(1~10个D4Z4拷贝数),存在等位序列4qA;FSHD2患者4q35-EcoRⅠ片段正常,存在相关基因致病突变(SMCHD1、DNMT3B和LRIF1);靶向CpG甲基化检测可以获取4q35-D4Z4序列的表观遗传学诊断信息。
鉴别诊断
1.肢带型肌营养不良 多于10~20岁起病,主要表现为对称性肩胛带和骨盆带肌无力和萎缩,可伴假性肌肥大,较少累及面肌,病程缓慢进展,常染色体显性或隐性遗传,肌电图提示肌源性损害,缺陷蛋白的肌肉免疫病理检测可辅助诊断,肌肉MRI及基因检测有助于鉴别及分类。
2.强直性肌营养不良症 常显遗传病,多为成年后发病,临床表现为四肢肌无力、萎缩及强直,多为远端型起病,屈颈无力,常有面部表情肌受累,伴肌强直及肌僵硬,典型者有“斧头脸”、“鹅颈”、握拳松开动作迟缓及叩击性肌球,肌电图出现肌强直放电,DMPK基因检测动态突变可以确诊。
3.线粒体肌病 起病年龄不定,以躯干肌及四肢近端不耐受疲劳为主要特征,活动后肌无力加重,休息后好转,可伴眼外肌麻痹,血乳酸、丙酮酸升高,肌电图以肌源性损害为主,光镜病理可见破碎红肌纤维(ragged red fibers, RRF/MGT染色)及蓝纤维(COX/SDH染色),电镜示肌膜下和肌原纤维间大量异常线粒体堆积。人体组织(肌肉、毛囊、脱落细胞等)较血液的线粒体DNA突变检测确诊率为高。
4.多发性肌炎 急性或亚急性起病,表现为渐进性、对称性四肢近端、颈肌及咽肌无力伴肌肉压痛,伴血清肌酶升高,肌电图示活动期肌源性损害,肌活检示肌细胞间或肌细胞内炎性浸润,肌炎抗体谱检测有助于分型,对免疫抑制治疗有效。
治疗
目前尚无特效药物,治疗策略分为两类:
1.增加肌肉体积或力量 适量有氧运动有益,不仅改善心血管健康,还可以增加力量。力量训练包括渐进的负荷训练、以肘部屈肌和踝关节背屈肌为重点的动态和等距训练。针对上肢抬举困难和足下垂,可应用矫形器或者手术改善。踝-足矫形器或膝-踝-足矫形器的使用可改善足下垂患者的活动能力和避免跌倒。肩胛骨固定手术可在一定程度上改善肩关节功能、肩关节活动度或肩胛骨的疼痛,但要注意肺部的并发症。
2.靶向治疗 近几年,在小分子化合物、反义寡核苷酸 (antisense oligonucleotides,ASO)、新型基因编辑等研究取得一定进展,包括针对DUX4的分子抑制或下游靶点,以沉默DUX4基因或者阻断DUX4蛋白。小分子化合物包括β2肾上腺素能受体激动剂、BET蛋白BRD4抑制剂、p38 MAPK抑制剂Losmapimod(洛司莫德)、P300组蛋白乙酰转移酶抑制剂等,其中Losmapimod的II期临床试验结果虽然证实了安全性和有效性,但最近Ⅲ期临床试验未能达到主要和次要终点。AOC 1020已获得美国食品药品监管局(FDA)的快速通道和孤儿药认定资格,处于1/2期临床研究,重点评估静脉给药的安全性和人体耐受性。
诊疗流程 (图25-4)
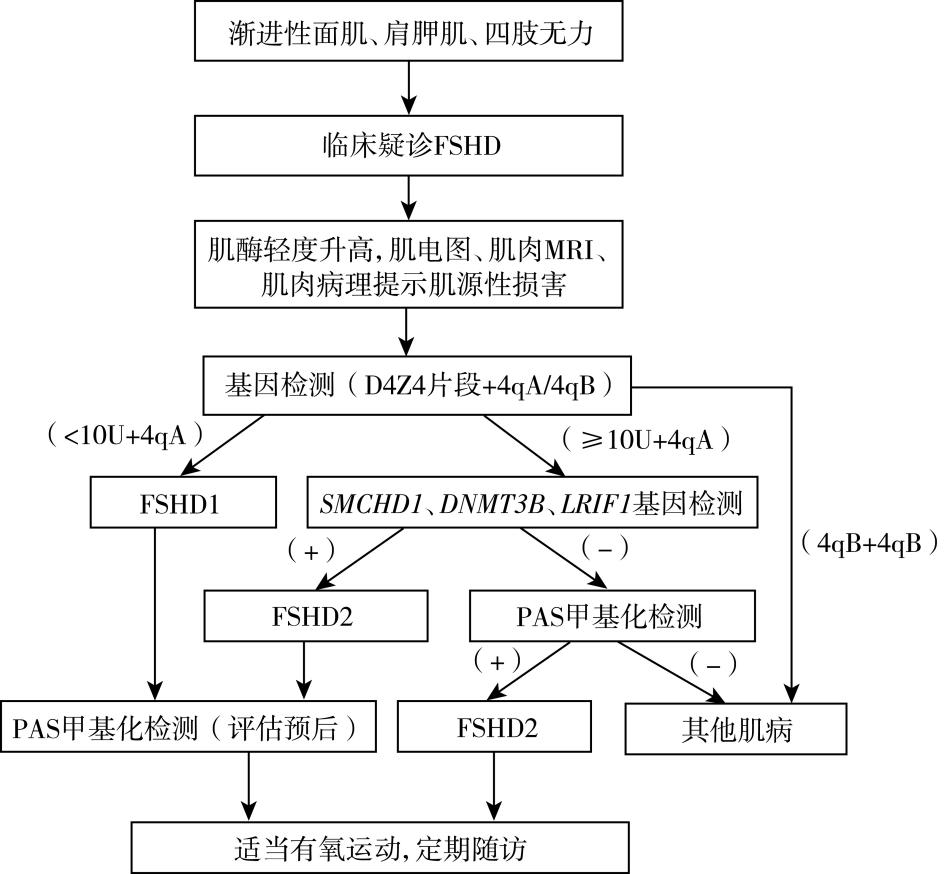 图25-4 面肩肱型肌营养不良症诊疗流程
图25-4 面肩肱型肌营养不良症诊疗流程
参考文献
[1]SORREL-DEJERINE Y, FARDEAU M. Naissance et métamorphoses de la myopathie atrophique progressive de Landouzy et Déjerine [Birth and metamorphosis of Landouzy-Dejerine progressive atrophic myopathy]. Rev Neurol (Paris),1982,138(12):1041-1051.
[2]TYLER FH, STEPHENS FE. Studies in disorders of muscle. Ⅱ clinical manifestations and inheritance of facioscapulohumeral dystrophy in a large family. Ann Intern Med,1950,32(4):640-660.
[3]DEENEN JC, ARNTS H, VAN DER MAAREL SM, et al. Population-based incidence and prevalence of facioscapulohumeral dystrophy. Neurology,2014,83(12):1056-1059.
[4]WANG ZQ, QIU LL, LIN MT, et al. Prevalence and disease progression of genetically-confirmed facioscapulohumeral muscular dystrophy type 1 (FSHD1) in China between 2001 and 2020: a nationwide population-based study. Lancet Reg Health West Pac,2022,18:100323.
[5]QIU LL, LIN XD, XU GR, et al. A novel start codon variant in SMCHD1 from a Chinese family causes facioscapulohumeral muscular dystrophy type 2. Chin Med J (Engl),2021,134(22):2753-2755.
[6]HAMANAKA K, ŠIKROVÁ D, MITSUHASHI S, et al. Homozygous nonsense variant in LRIF1 associated with facioscapulohumeral muscular dystrophy. Neurology,2020,94(23):e2441-e2447.
[7]LEMMERS RJLF, TAWIL R, PETEK LM, et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat Genet,2012,44(12):1370-1374.
[8]VAN DEN BOOGAARD ML, LEMMERS RJLF, BALOG J, et al. Mutations in DNMT3B modify epigenetic repression of the D4Z4 Repeat and the penetrance of facioscapulohumeral dystrophy. Am J Hum Genet,2016,98(5):1020-1029.
[9]LEMMERS RJ, VAN DER VLIET PJ, KLOOSTER R, et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science,2010,329(5999):1650-1653.
[10]LIN F, HE JJ, LIN XD, et al. A large cohort study confirming that specific haplotype 4A161PAS is exclusively associated with the Chinese FSHD1. Clin Genet,2016,90(6):558-559.
[11]MUL K. Facioscapulohumeral muscular dystrophy. Continuum (Minneap Minn),2022,28(6):1735-1751.
[12]KATZ NK, HOGAN J, DELBANGO R, et al. Predictors of functional outcomes in patients with facioscapulohumeral muscular dystrophy. Brain,2021,144(11):3451-3460.
[13]QIU LL, CHEN L, ZHENG FZ, et al. Wheelchair use in genetically confirmed FSHD1 from a large cohort study in Chinese population. Brain,2022,145(6):e51-e54.
[14]HE JJ, LIN XD, LIN F, et al. Clinical and genetic features of patients with facial-sparing facioscapulohumeral muscular dystrophy. Eur J Neurol,2018,25(2):356-364.
[15]LAMPERTI C, FABBRI G, VERCELLI L, et al. A standardized clinical evaluation of patients affected by facioscapulohumeral muscular dystrophy: The FSHD clinical score. Muscle Nerve,2010,42(2):213-217.
[16]RICCI G, RUGGIERO L, VERCELLI L, et al. A novel clinical tool to classify facioscapulohumeral muscular dystrophy phenotypes. J Neurol,2016,263(6):1204-1214.
[17]ZHENG FZ, QIU LL, CHEN L, et al. Association of 4qA-Specific distal D4Z4 hypomethylation with disease severity and progression in facioscapulohumeral muscular dystrophy. Neurology,2023,101(3):e225-e237.
[18]QIU LL, YE ZX, LIN L, et al. Clinical and genetic features of somatic mosaicism in facioscapulohumeral dystrophy. J Med Genet,2020,57(11):777-785.
[19]ZHENG FZ, QIU LL, CHEN L, et al. An epigenetic basis for genetic anticipation in facioscapulohumeral muscular dystrophy type 1. Brain,2023,146(12):e107-e110.
[20]DAHLQVIST JR, ANDERSEN G, KHAWAJAZADA T, et al. Relationship between muscle inflammation and fat replacement assessed by MRI in facioscapulohumeral muscular dystrophy. J Neurol,2019,266(5):1127-1135.
[21]EFTHYMIOU S, LEMMERS RJLF, VISHNU VY, et al. Optical genome mapping for the molecular diagnosis of facioscapulohumeral muscular dystrophy: advancement and challenges. Biomolecules,2023,13(11):1567.
[22]BUTTERFIELD RJ, DUNN DM, DUVAL B, et al. Deciphering D4Z4 CpG methylation gradients in fascioscapulohumeral muscular dystrophy using nanopore sequencing. Genome Res,2023,33(9):1439-1454.
[23]GIARDINA E, CAMAÑO P, BURTON-JONES S, et al. Best practice guidelines on genetic diagnostics of facioscapulohumeral muscular dystrophy: update of the 2012 guidelines. Clin Genet,2024,106(1):13-26.
[24]EREN İ, ERŞEN A, BIRSEL O, et al. Functional outcomes and complications following scapulothoracic arthrodesis in patients with facioscapulohumeral dystrophy. JBJS,2020,102(3):237-244.
[25]TIHAYA MS, MUL K, BALOG J, et al. Facioscapulohumeral muscular dystrophy: the road to targeted therapies. Nat Rev Neurol,2023,19(2):91-108.
[26]TAWIL R, WAGNER KR, HAMEL JI, et al. Safety and efficacy of losmapimod in facioscapulohumeral muscular dystrophy (ReDUX4): a randomised, double-blind, placebo-controlled phase 2b trial. Lancet Neurol,2024,23(5):477-486.